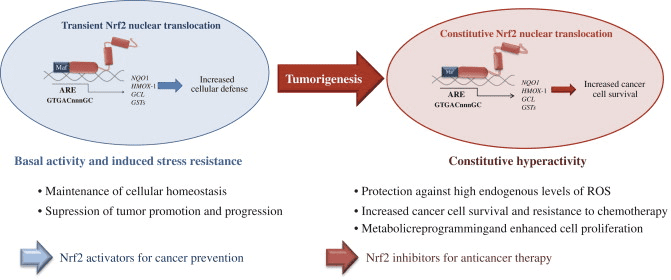
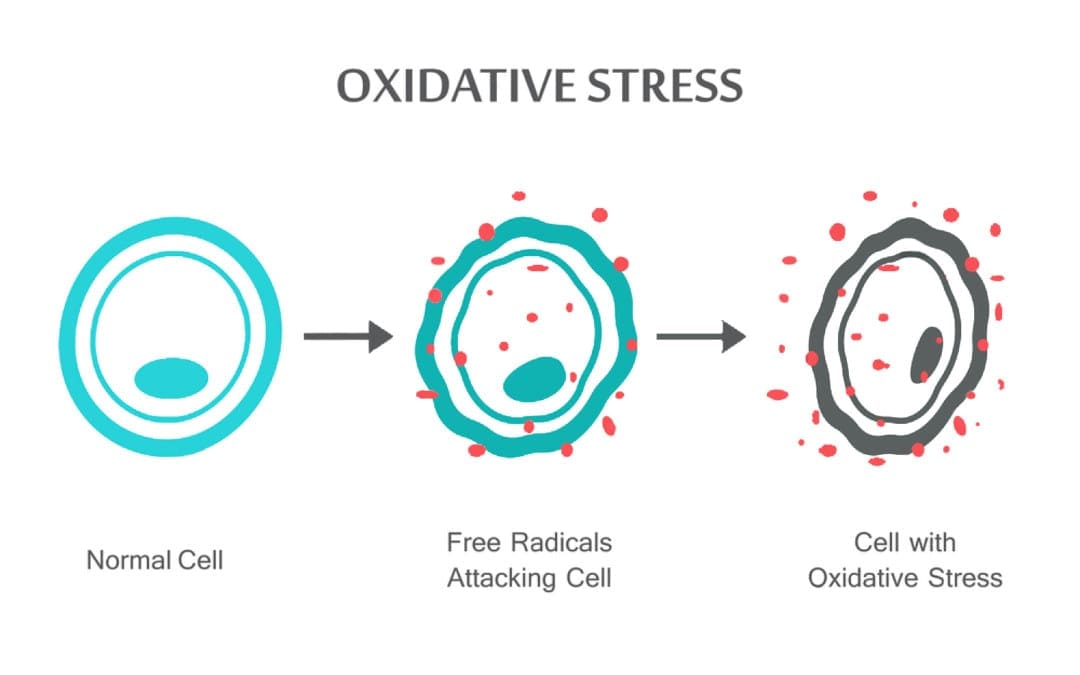
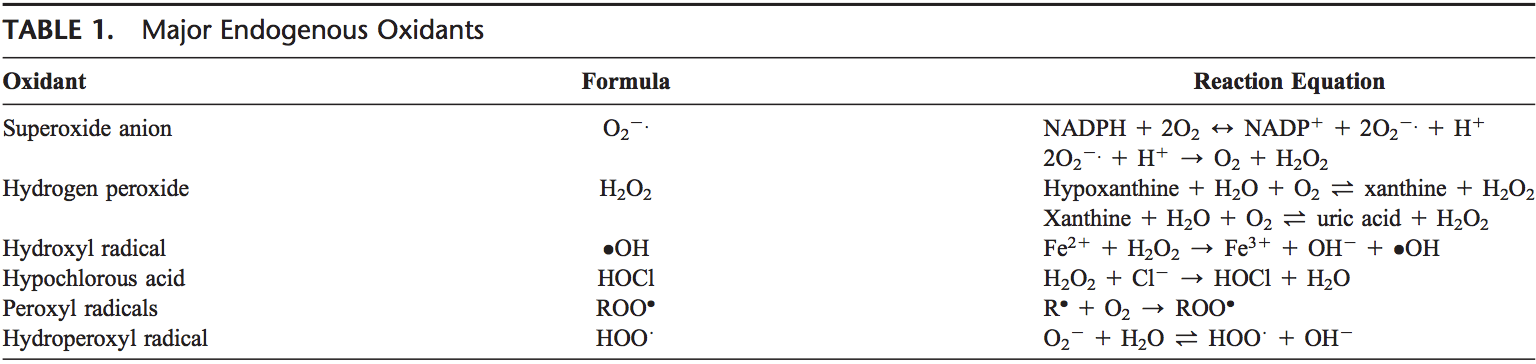
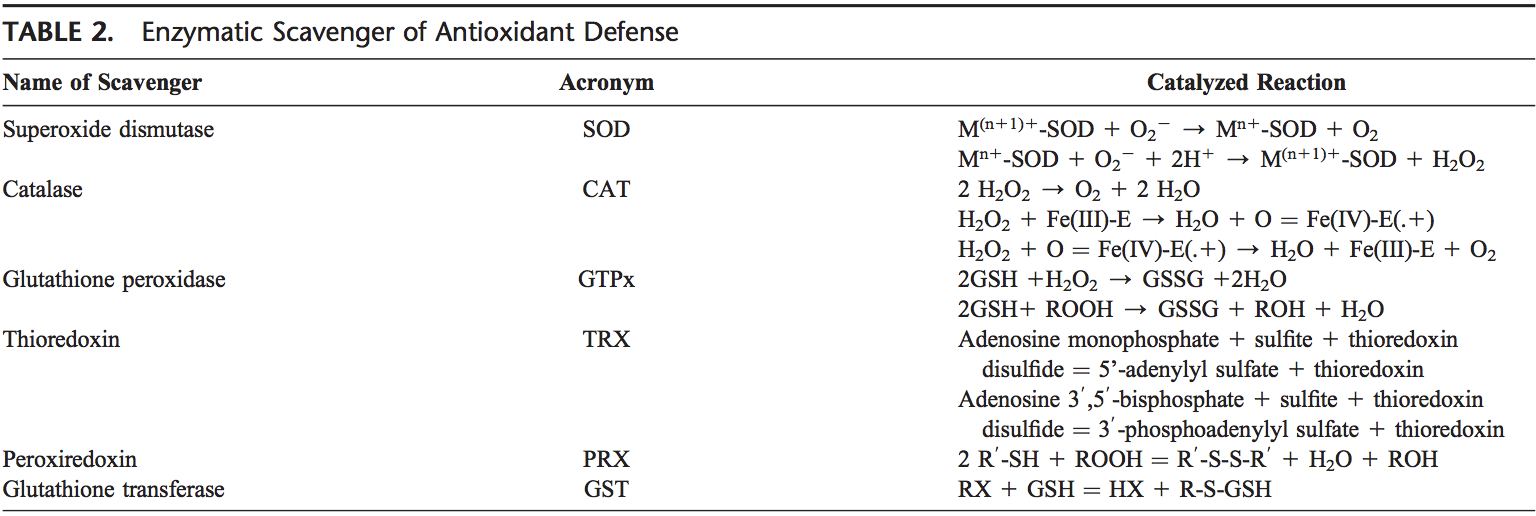
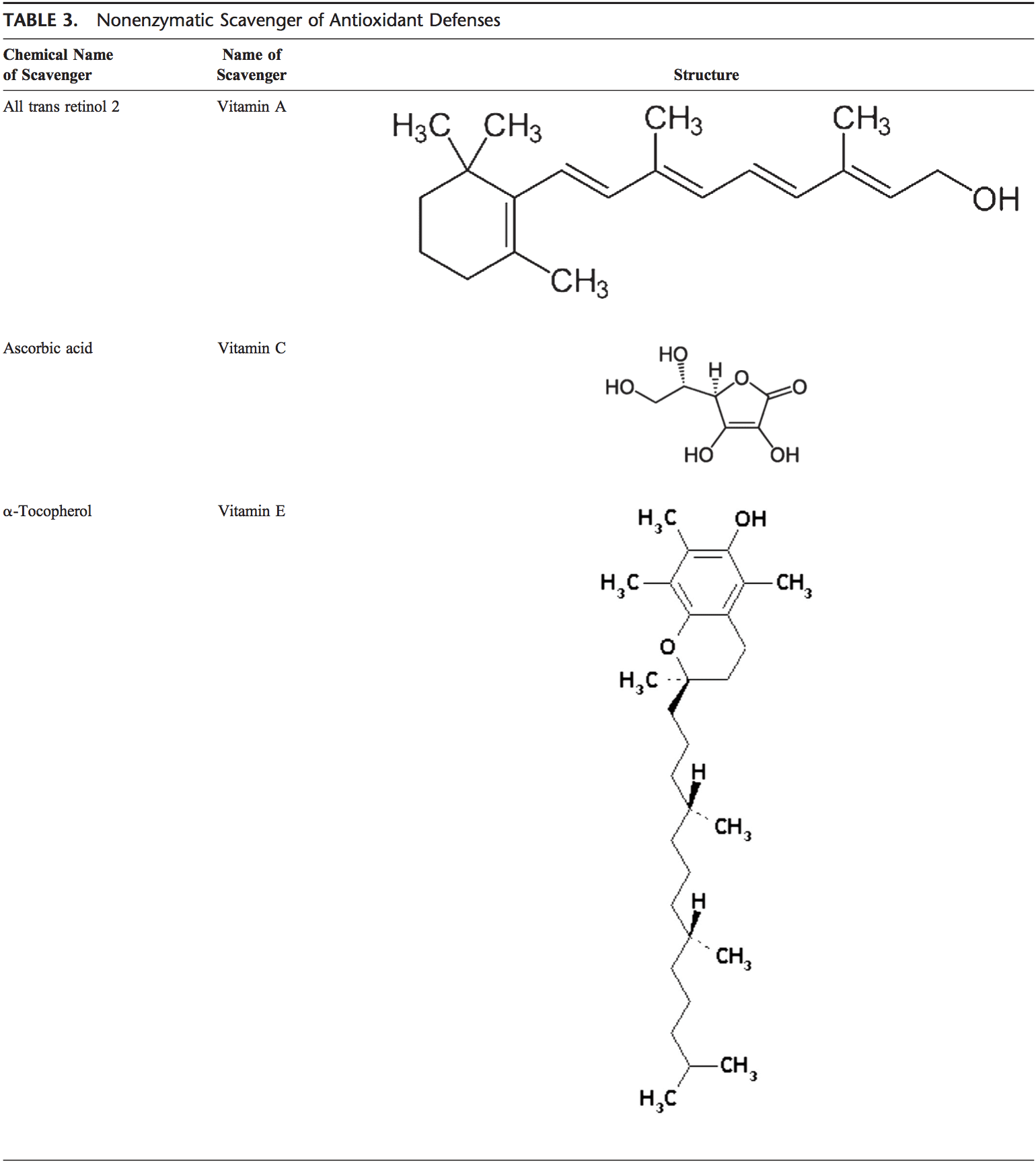
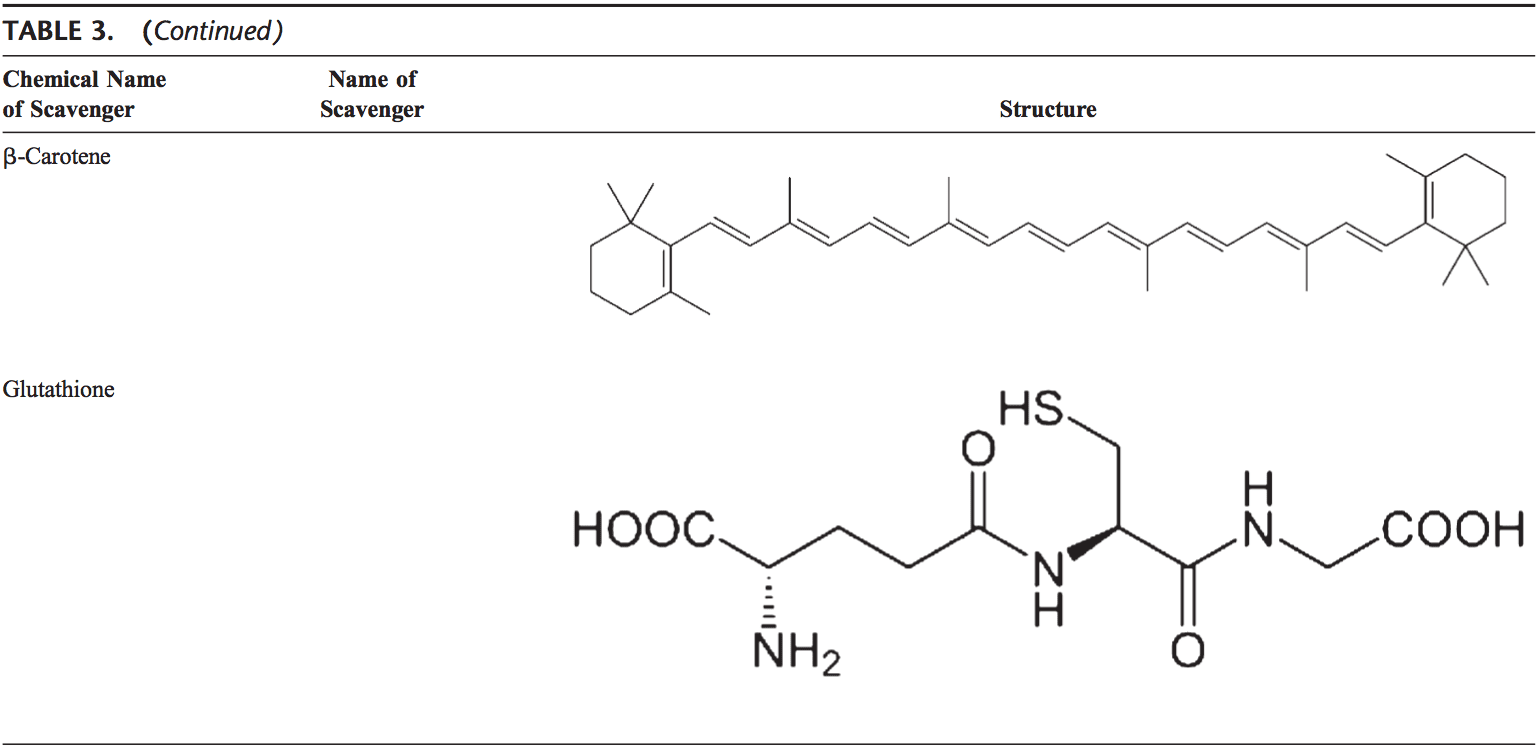
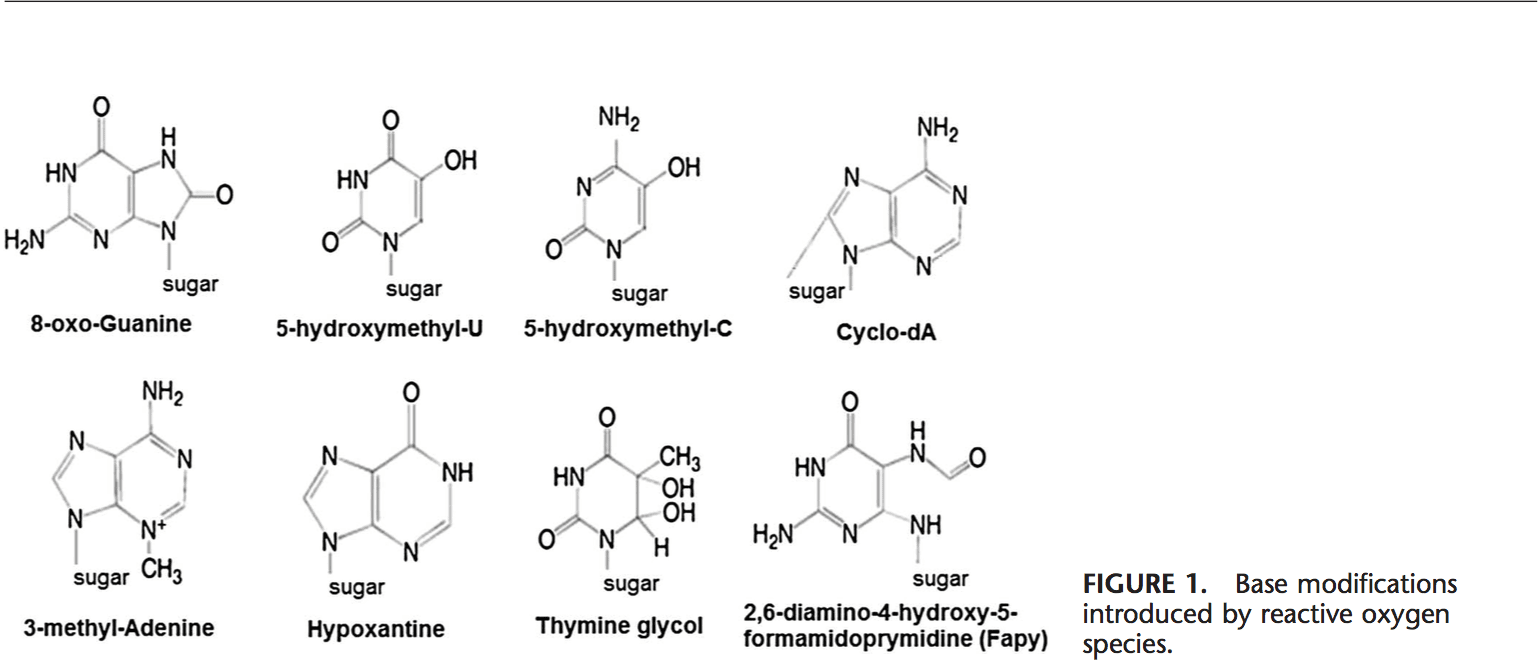
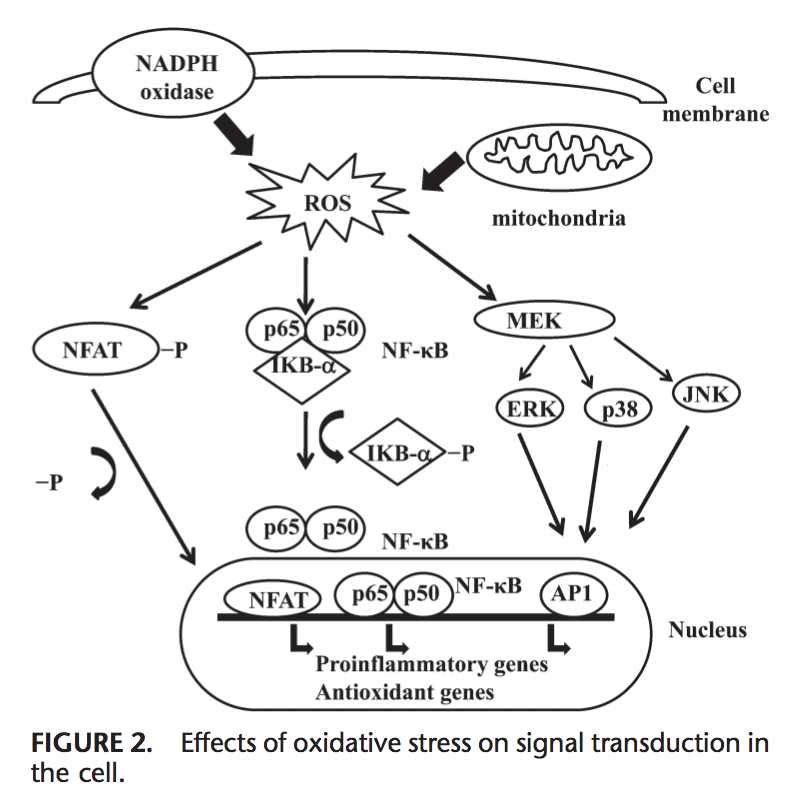
Rygklinik Oxidativ Stress Kiropraktik og Funktionel Medicin Team. Oxidativ stress er defineret som en forstyrrelse i balancen mellem produktionen af reaktivt ilt (frie radikaler) og antioxidantforsvar. Det er med andre ord en ubalance mellem produktionen af frie radikaler og kroppens evne til at modvirke eller afgifte de skadelige effekter gennem neutralisering af antioxidanter. Oxidativ stress fører til mange patofysiologiske tilstande i kroppen. Disse omfatter neurodegenerative sygdomme, dvs. Parkinsons sygdom, Alzheimers sygdom, genmutationer, kræftformer, kronisk træthedssyndrom, skrøbeligt X-syndrom, hjerte- og blodkarsygdomme, aterosklerose, hjertesvigt, hjerteanfald og inflammatoriske sygdomme. Oxidation sker under en række omstændigheder:
cellerne bruger glukose til at lave energi
immunsystemet bekæmper bakterier og skaber betændelse
kroppene afgifter forurenende stoffer, pesticider og cigaretrøg
Der foregår millioner af processer i vores kroppe på ethvert givet tidspunkt, som kan resultere i oxidation. Her er et par symptomer:
Træthed
Hukommelsestab og eller hjernetåge
Muskel- og eller ledsmerter
Rynker sammen med gråt hår
Nedsat syn
Hovedpine og følsomhed over for støj
Modtagelighed for infektioner
At vælge økologiske fødevarer og undgå toksiner i dit miljø gør en stor forskel. Dette, sammen med at reducere stress, kan være gavnligt til at reducere oxidation.
Oxidanter produceres generelt på en kontrolleret måde for at regulere væsentlige processer i den menneskelige krop, herunder celledeling, inflammation, immunfunktion, autofagi og stressrespons. Den ukontrollerede produktion af disse oxidanter kan dog bidrage til oxidativt stress, hvilket kan påvirke cellulær funktion, hvilket fører til udvikling af toksicitet, kronisk sygdom og cancer. Den menneskelige krops beskyttende antioxidantmekanismer reguleres af en række vitale veje, der styrer cellens reaktion på oxidanter. Den nukleare faktor erythroid 2-relaterede faktor, ellers kendt som Nrf2, er en ny regulator af cellulær modstand mod oxidanter. Formålet med artiklen nedenfor er at diskutere og demonstrere Nrf2's nye rolle i mitokondriel funktion.
Abstrakt
Den transkriptionsfaktor NF-E2 p45-relaterede faktor 2 (Nrf2; gennavn NFE2L2) tillader tilpasning og overlevelse under stresstilstande ved at regulere genekspressionen af forskellige netværk af cytobeskyttende proteiner, herunder antioxidant-, anti-inflammatoriske og afgiftningsenzymer. som proteiner, der hjælper med reparation eller fjernelse af beskadigede makromolekyler. Nrf2 har en afgørende rolle i opretholdelsen af cellulær redox-homeostase ved at regulere biosyntesen, udnyttelsen og regenereringen af glutathion, thioredoxin og NADPH og ved at kontrollere produktionen af reaktive oxygenarter af mitokondrier og NADPH-oxidase. Under homøostatiske forhold påvirker Nrf2 mitokondriemembranpotentialet, fedtsyreoxidation, tilgængeligheden af substrater (NADH og FADH2/succinat) til respiration og ATP-syntese. Under betingelser med stress eller vækstfaktorstimulering modvirker aktivering af Nrf2 den øgede produktion af reaktive oxygenarter i mitokondrier via transkriptionel opregulering af afkoblingsprotein 3 og påvirker mitokondriel biogenese ved at opretholde niveauerne af nuklear respiratorisk faktor 1 og peroxisomproliferator-aktiveret receptor ? coactivator 1?, såvel som ved at fremme purin-nukleotidbiosyntese. Farmakologiske Nrf2-aktivatorer, såsom det naturligt forekommende isothiocyanat sulforaphane, hæmmer oxidant-medieret åbning af mitokondrielle permeabilitetsovergangsporer og mitokondriel hævelse. Mærkeligt nok blev det fundet, at en syntetisk 1,4-diphenyl-1,2,3-triazolforbindelse, oprindeligt designet som en Nrf2-aktivator, fremmer mitofagi og derved bidrager til den overordnede mitokondrielle homeostase. Således er Nrf2 en fremtrædende aktør i at understøtte mitokondriernes strukturelle og funktionelle integritet, og denne rolle er særligt afgørende under stresstilstande.
Nrf2 har en afgørende rolle i at opretholde cellulær redox-homeostase.
Nrf2 påvirker mitokondrielle membranpotentiale og ATP-syntese.
Nrf2 påvirker mitokondriel fedtsyreoxidation.
Nrf2 understøtter mitokondriernes strukturelle og funktionelle integritet.
Nrf2-aktivatorer har gavnlige virkninger, når mitokondriefunktionen er kompromitteret.
Introduktion
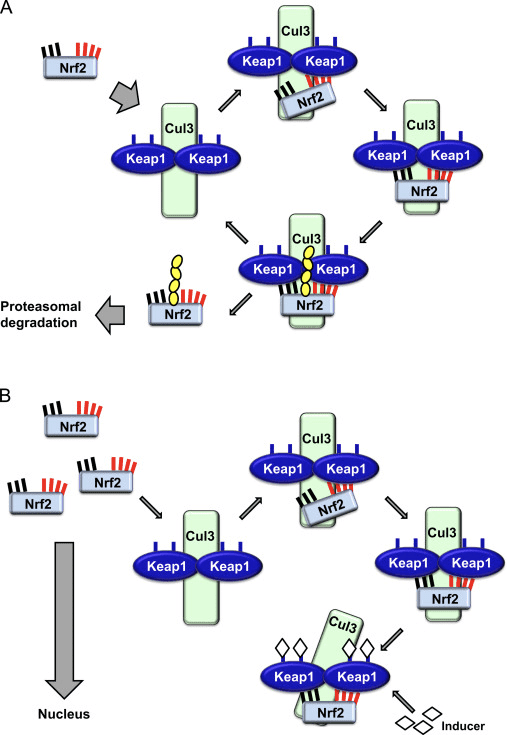
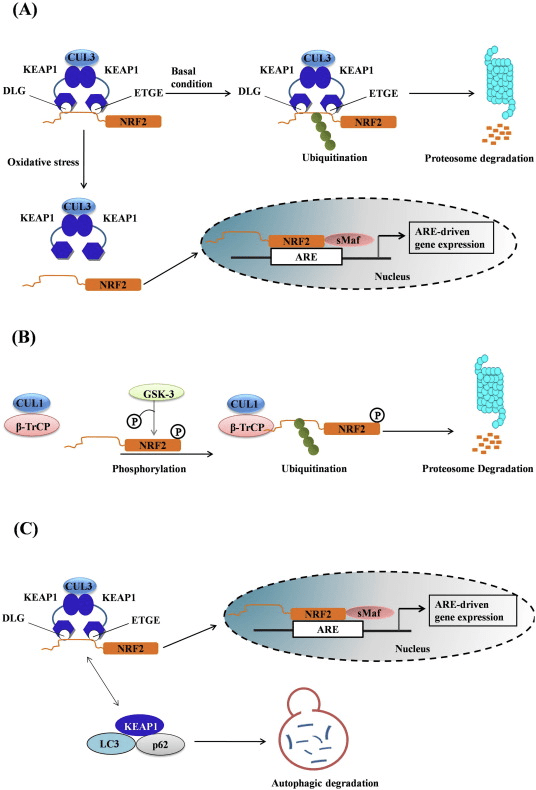
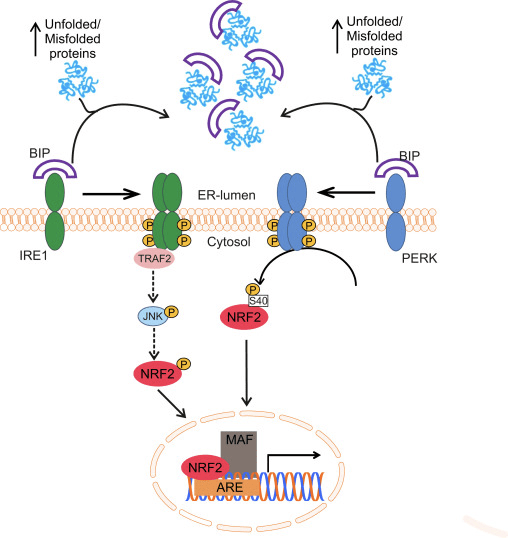
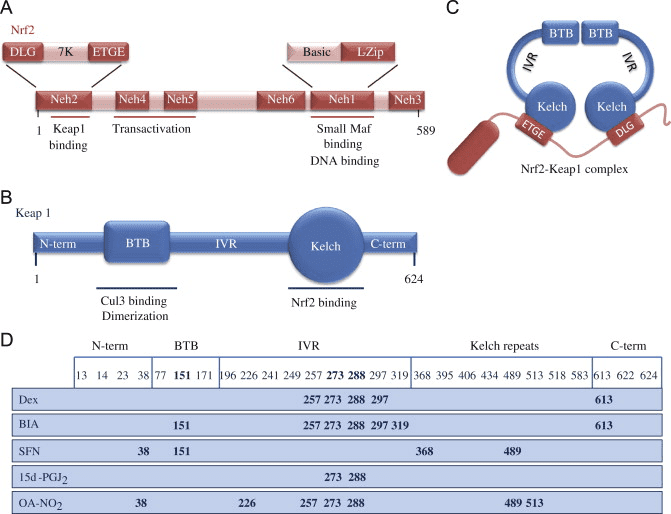
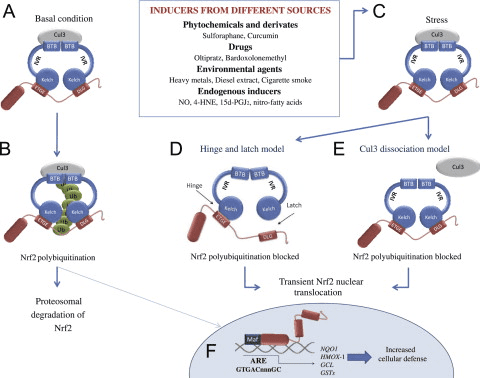
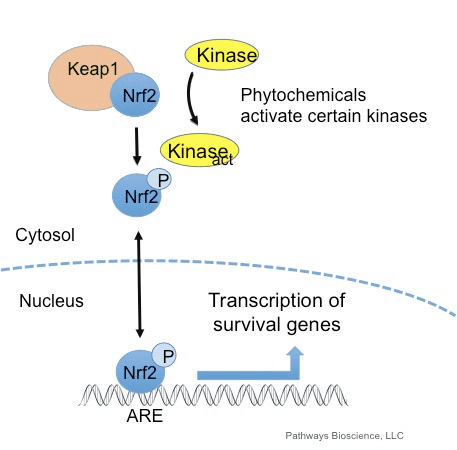
Transkriptionsfaktoren NF-E2 p45-relateret faktor 2 (Nrf2; gennavn NFE2L2) regulerer ekspressionen af netværk af gener, der koder for proteiner med forskellige cytobeskyttende aktiviteter. Nrf2 selv styres primært på niveauet af proteinstabilitet. Under basale forhold er Nrf2 et kortlivet protein, der udsættes for kontinuerlig ubiquitinering og proteasomal nedbrydning. Der er tre kendte ubiquitin-ligasesystemer, der bidrager til nedbrydningen af Nrf2. Historisk set var den første negative regulator af Nrf2, der blev opdaget, Kelch-lignende ECH-associeret protein 1 (Keap1) [1], et substratadapterprotein for Cullin 3 (Cul3)/Rbx1 ubiquitinligase [2], [3], [ 4]. Keap1 bruger en meget effektiv cyklisk mekanisme til at målrette Nrf2 til ubiquitinering og proteasomal nedbrydning, hvor Keap1 kontinuerligt regenereres, hvilket tillader cyklussen at fortsætte (fig. 1A) [5]. Nrf2 udsættes også for nedbrydning medieret af glykogensyntasekinase (GSK)3/a-TrCP-afhængig Cul1-baseret ubiquitinligase [6], [7]. Senest blev det rapporteret, at under tilstande med endoplasmatisk retikulumstress, bliver Nrf2 ubiquitineret og nedbrudt i en proces medieret af E3 ubiquitin-ligasen Hrd1 [8].
Figur 1 Den cykliske sekventielle bindings- og regenereringsmodel for Keap1-medieret nedbrydning af Nrf2. (A) Nrf2 binder sekventielt til en fri Keap1 dimer: først gennem dets højaffinitet ETGE (røde stifter) bindingsdomæne og derefter gennem dets lavaffinitet DLG (sorte stifter) bindingsdomæne. I denne konformation af proteinkomplekset gennemgår Nrf2 ubiquitinering og er målrettet mod proteasomal nedbrydning. Fri Keap1 regenereres og er i stand til at binde sig til nyligt oversat Nrf2, og cyklussen begynder igen.(B) Inducere (hvide diamanter) reagerer med sensorcysteiner af Keap1 (blå sticks), hvilket fører til en konformationsændring og forringet substratadapteraktivitet. Fri Keap1 regenereres ikke, og den nyligt syntetiserede Nrf2 akkumuleres og translokeres til kernen.
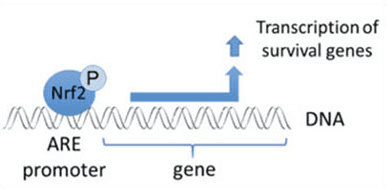
Ud over at fungere som et ubiquitin-ligase-substratadapterprotein er Keap1 også sensoren for en bred vifte af småmolekylære aktivatorer af Nrf2 (kaldet inducere) [9]. Inducere blokerer cyklussen af Keap1-medieret nedbrydning af Nrf2 ved kemisk at modificere specifikke cysteinrester i Keap1 [10], [11] eller ved direkte at forstyrre Keap1:Nrf2-bindingsgrænsefladen [12], [13]. Som følge heraf nedbrydes Nrf2 ikke, og transkriptionsfaktoren akkumuleres og translokeres til kernen (Fig. 1B), hvor den danner en heterodimer med et lille Maf-protein; binder til antioxidant-respons-elementer, de opstrøms regulatoriske områder af dets målgener; og initierer transkription [14], [15], [16]. Batteriet af Nrf2-mål omfatter proteiner med forskellige cytobeskyttende funktioner, herunder enzymer af xenobiotisk metabolisme, proteiner med antioxidant- og antiinflammatoriske funktioner og proteasomale underenheder, samt proteiner, der regulerer cellulær redox-homeostase og deltager i intermediær metabolisme.
Nrf2: en Master Regulator of Cellular Redox Homeostase
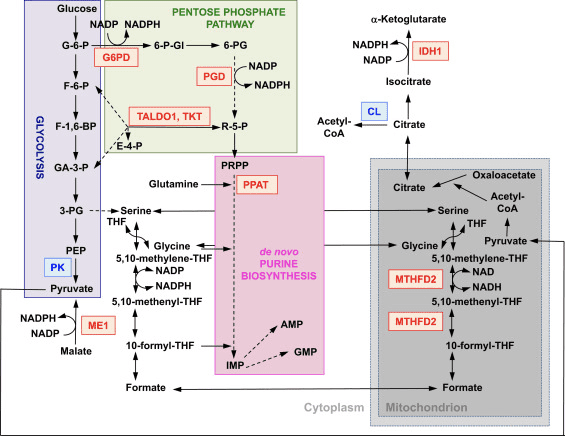
Funktionen af Nrf2 som en master regulator af cellulær redox homeostase er bredt anerkendt. Genekspressionen af både de katalytiske og de regulatoriske underenheder af β-glutamylcysteinligase, enzymet der katalyserer det hastighedsbegrænsende trin i biosyntesen af reduceret glutathion (GSH), er direkte reguleret af Nrf2 [17]. xCT-underenheden af system xc-, som importerer cystin til celler, er også et direkte transkriptionelt mål for Nrf2 [18]. I cellen gennemgår cystin omdannelse til cystein, en forløber for biosyntesen af GSH. Ud over sin rolle i GSH-biosyntese giver Nrf2 midlerne til opretholdelse af glutathion i sin reducerede tilstand ved den koordinerede transkriptionelle regulering af glutathionreduktase 1 [19], [20], som reducerer oxideret glutathion til GSH ved hjælp af reducerende ækvivalenter fra NADPH . Den nødvendige NADPH leveres af fire primære NADPH-genererende enzymer, æblesyreenzym 1 (ME1), isocitratdehydrogenase 1 (IDH1), glucose-6-phosphatdehydrogenase (G6PD) og 6-phosphogluconatdehydrogenase (PGD), som alle er transskriptionelt reguleret delvist af Nrf2 (fig. 2) [21], [22], [23], [24]. Mærkeligt nok regulerer Nrf2 også den inducerbare genekspression af de cytosoliske, mikrosomale og mitokondrielle former af aldehyddehydrogenase [25], som bruger NAD(P)+ som en cofaktor, hvilket giver anledning til NAD(P)H. Faktisk er niveauerne af NADPH og NADPH/NADP+-forholdet lavere i embryonale fibroblaster isoleret fra Nrf2-knockout (Nrf2-KO) mus sammenlignet med celler fra deres vildtype (WT) modstykker, og NADPH-niveauerne falder ved Nrf2-knockdown i cancercellelinjer med konstitutivt aktiv Nrf2 [26]. Som forventet er niveauerne af GSH lavere i celler, hvor Nrf2 er blevet forstyrret; omvendt fører Nrf2-aktivering ved genetiske eller farmakologiske midler til GSH-opregulering [27], [28], [29]. Det er vigtigt, at Nrf2 også regulerer genekspressionen af thioredoxin [30], [31], [32], thioredoxin reduktase 1 [28], [29], [32], [33] og sulfiredoxin [34], som er essentielle til reduktion af oxiderede proteinthioler.
Figur 2 Nrf2's rolle i metabolismen af hurtigt prolifererende celler. Nrf2 er en positiv regulator af gener, der koder for enzymer i både den oxidative arm [dvs. glucose-6-phosphat dehydrogenase (G6PD) og 6-phosphogluconate dehydrogenase (PGD)] og den ikke-oxidative arm [dvs. transaldolase 1 (TALDO1) og transketolase ( TKT)] af pentosephosphatvejen. G6PD og PGD genererer NADPH. Nrf2 regulerer også genekspressionen af de to andre NADPH-genererende enzymer, æblesyreenzym 1 (ME1) og isocitratdehydrogenase 1 (IDH1). Genekspressionen af phosphoribosyl pyrophosphate amidotransferase (PPAT), som katalyserer adgangen til de novo purin biosyntesevejen, er også positivt reguleret af Nrf2, ligesom ekspressionen af methylentetrahydrofolat dehydrogenase 2 (MTHFD2), en mitokondriel rolle i enzymet med en kritisk rolle tilvejebringelse af en-carbonenheder til de novo purinbiosyntese. Pyruvatkinase (PK) er negativt reguleret af Nrf2 og forventes at favorisere opbygningen af glykolytiske mellemprodukter og sammen med G6PD metabolitkanalisering gennem pentosephosphatvejen og syntesen af nukleinsyrer, aminosyrer og fosfolipider. Nrf2 regulerer negativt genekspressionen af ATP-citratlyase (CL), hvilket kan øge tilgængeligheden af citrat til mitokondriel udnyttelse eller (gennem isocitrat) for IDH1. Rød og blå angiver henholdsvis positiv og negativ regulering. Mitokondriet er vist i gråt. Metabolitforkortelser: G-6-P, glucose 6-phosphat; F-6-P, fructose-6-phosphat; F-1,6-BP, fructose-1,6-bisphosphat; GA-3-P, glyceraldehyd-3-phosphat; 3-PG, 3-phosphoglycerat; PEP, phosphoenolpyruvat; 6-P-Gl, 6-phosphogluconolacton; 6-PG, 6-phosphogluconat; R-5-P, ribulose-5-phosphat; PRPP, 5-phosphoribosyl-a-1-pyrophosphat; THF, tetrahydrofolat; IMP, inosinmonophosphat; AMP, adenosinmonophosphat; GMP, guanosinmonofosfat.
I betragtning af Nrf2's afgørende rolle som en masterregulator af cellulær redoxhomeostase er det ikke overraskende, at sammenlignet med WT-celler er niveauerne af reaktive oxygenarter (ROS) højere i celler, hvor Nrf2 er blevet forstyrret (Nrf2-KO) [35]. Denne forskel er især slående ved udfordring med midler, der forårsager oxidativ stress. Desuden er celler, der mangler Nrf2, meget mere følsomme over for toksiciteten af oxidanter af forskellige typer og kan ikke beskyttes af Nrf2-inducere, som under de samme betingelser giver effektiv og langvarig beskyttelse til WT-celler [29], [36] , [37]. Ud over den overordnede cellulære redox-homeostase er Nrf2 også kritisk for opretholdelsen af mitokondriel redox-homeostase. Sammenlignet med WT er den samlede mitokondrielle NADH-pulje signifikant øget i Keap1-KO og dramatisk reduceret i Nrf2-KO-celler [35].
Ved hjælp af levende cellebilleddannelse overvågede vi for nylig hastighederne for ROS-produktion i primære glioneuronale cokulturer og hjernevævsskiver isoleret fra WT, Nrf2-KO eller Keap1-knockdown (Keap1-KD) mus [38]. Som forventet var hastigheden af ROS-produktion hurtigere i Nrf2-KO-celler og væv sammenlignet med deres WT-modstykker. Vi gjorde dog den uventede observation, at sammenlignet med WT har Keap1-KD-celler også højere ROS-produktion, selvom størrelsen af forskellen mellem WT- og Keap1-KD-genotyperne var mindre end den mellem WT og Nrf2-KO . Vi analyserede derefter mRNA-niveauerne af NOX2 og NOX4, de katalytiske underenheder af de to NADPH-oxidase (NOX) isoformer, der er blevet impliceret i hjernepatologi, og fandt ud af, at NOX2 er dramatisk øget under forhold med Nrf2-mangel, hvorimod NOX4 opreguleres, når Nrf2 er konstitutivt aktiveret, dog i mindre omfang. Kvantitativt svarer størrelsen af opregulering i celler og væv fra mutantmusene til de tilsvarende stigninger i ROS-produktion [38]. Interessant nok regulerer Nrf2 ikke kun NADPH-oxidase, men ROS produceret af NADPH-oxidase kan aktivere Nrf2, som vist i lungeepitelceller og kardiomyocytter [39], [40]. Ydermere har en meget nylig undersøgelse vist, at den NADPH-oxidaseafhængige aktivering af Nrf2 udgør en vigtig endogen mekanisme til beskyttelse mod mitokondriel skade og celledød i hjertet under kronisk trykoverbelastning [41].
Ud over den katalytiske aktivitet af NADPH-oxidase er mitokondriel respiration en anden vigtig intracellulær kilde til ROS. Ved brug af den mitokondriespecifikke probe MitoSOX har vi undersøgt bidraget af ROS af mitokondriel oprindelse til den samlede ROS-produktion i primære glioneuronale cokulturer isoleret fra WT, Nrf2-KO eller Keap1-KD mus [38]. Som forventet havde Nrf2-KO-celler højere hastigheder af mitokondriel ROS-produktion end WT. I overensstemmelse med resultaterne for den samlede ROS-produktion var hastighederne for mitokondriel ROS-produktion i Keap1-KD også højere sammenlignet med WT-celler. Det er vigtigt, at blokering af kompleks I med rotenon forårsagede en dramatisk stigning i mitokondriel ROS-produktion i både WT- og Keap1-KD-celler, men havde ingen effekt i Nrf2-KO-celler. I modsætning til den forventede stigning i mitokondriel ROS-produktion i WT-celler efter tilsætning af pyruvat (for at øge tilgængeligheden af NADH, øge mitokondriel membranpotentiale og normalisere respiration), faldt produktionen af ROS i Nrf2-KO-celler. Tilsammen tyder disse resultater stærkt på, at i fravær af Nrf2: (i) aktiviteten af kompleks I er svækket, (ii) den forringede aktivitet af kompleks I skyldes begrænsning af substrater, og (iii) den svækkede aktivitet af kompleks I er en af hovedårsagerne til den øgede mitokondrielle ROS-produktion, muligvis på grund af omvendt elektronstrøm fra kompleks II.
Nrf2 påvirker mitokondriel membranpotentiale og respiration
Det mitokondrielle membranpotentiale (??m) er en universel indikator for mitokondriel sundhed og cellens metaboliske tilstand. I en sund celle opretholdes ??m af den mitokondrielle respiratoriske kæde. Interessant nok har en stabil isotopmærkning med aminosyrer i kulturbaseret proteomisk undersøgelse i den østrogenreceptor-negative ikke-tumorogene humane brystepitel MCF10A-cellelinje vist, at den mitokondrielle elektrontransportkæde-komponent NDUFA4 opreguleres af farmakologisk aktivering (af sulforaphane,) hvorimod genetisk opregulering af Nrf2 (ved Keap2 knockdown) fører til nedregulering af cytochrom c oxidase underenhederne COX1 og COX2I4 [1]. En undersøgelse af leverproteomet ved hjælp af todimensionel gelelektroforese og matrix-assisteret laserdesorptions-/ioniseringsmassespektrometri har fundet ud af, at Nrf42 regulerer ekspressionen af ATP-syntase-underenhed ? [2]. Derudover er mitokondrieproteinet DJ-43, som spiller en rolle i opretholdelsen af aktiviteten af kompleks I [1], blevet rapporteret at stabilisere Nrf44 [2], [45], selvom de neurobeskyttende virkninger af farmakologisk eller genetisk aktivering af Nrf46 er uafhængige af DJ-2 [1]. Konsekvenserne af disse observationer for mitokondriel funktion er dog ikke blevet undersøgt.
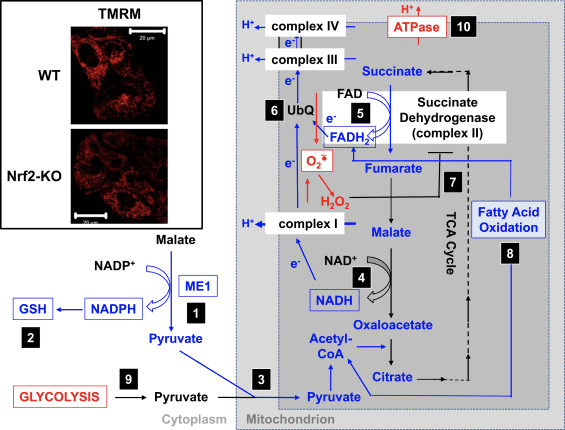
I overensstemmelse med den svækkede aktivitet af kompleks I under betingelser med Nrf2-mangel er den basale ??m lavere i Nrf2-KO museembryonale fibroblaster (MEF'er) og dyrkede primære glioneuronale celler sammenlignet med deres WT-modstykker (fig. 3, indsat) [35]. I modsætning hertil er den basale ??m højere, når Nrf2 er genetisk konstitutivt opreguleret (ved knockdown eller knockout af Keap1). Disse forskelle i ??m blandt genotyperne indikerer, at respirationen påvirkes af aktiviteten af Nrf2. Faktisk har evaluering af iltforbruget i den basale tilstand afsløret, at sammenlignet med WT er iltforbruget lavere i Nrf2-KO og Keap1-KO MEF'er med henholdsvis ~50 og ~35%.
Figur 3 Foreslået mekanisme for kompromitteret mitokondriefunktion under forhold med Nrf2-mangel. (1) De nedsatte niveauer af ME1, IDH1, G6PD og PGD resulterer i lavere NADPH-niveauer. (2) Niveauerne af GSH er også lave. (3) Den lave aktivitet af ME1 kan mindske puljen af pyruvat, der kommer ind i mitokondrierne. (4) Genereringen af NADH er langsommere, hvilket fører til nedsat aktivitet af kompleks I og øget mitokondriel ROS-produktion. (5) Reduktionen af FAD til FADH2 i mitokondrielle proteiner er også reduceret, hvilket sænker elektronstrømmen fra FADH2 til UbQ og ind i kompleks III. (6) Den langsommere dannelse af UbQH2 kan sænke enzymaktiviteten af succinatdehydrogenase. (7) De øgede niveauer af ROS kan yderligere hæmme aktiviteten af kompleks II. (8) Den lavere effektivitet af fedtsyreoxidation bidrager til den reducerede substrattilgængelighed for mitokondriel respiration. (9) Glykolyse forstærkes som en kompenserende mekanisme for den nedsatte ATP-produktion ved oxidativ fosforylering. (10) ATP-syntase fungerer omvendt for at opretholde ??m. Rød og blå angiver henholdsvis opregulering og nedregulering. Bokserne angiver tilgængeligheden af eksperimentelt bevis. Indsatsen viser billeder af mitokondrier af WT og Nrf2-KO kortikale astrocytter visualiseret af den potentiometriske fluorescerende probe tetramethylrhodamine methylester (TMRM; 25 nM). Målestok, 20 �m.
Disse forskelle i µm og respiration blandt genotyperne afspejles af hastigheden for udnyttelse af substrater til mitokondriel respiration. Anvendelse af substrater til tricarboxylsyre (TCA) cyklussen (malat/pyruvat, som igen øger produktionen af kompleks I-substratet NADH) eller methylsuccinat, et substrat for kompleks II, forårsager en trinvis stigning i ??m i både WT og Keap1-KD-neuroner, men stigningshastigheden er højere i Keap1-KD-celler. Endnu vigtigere er, at formerne af responsen på disse TCA-cyklussubstrater er forskellige mellem de to genotyper, hvorved den hurtige stigning i μm i Keap1-KD-celler efter substrattilsætning efterfølges af et hurtigt fald snarere end et plateau, hvilket tyder på et usædvanligt hurtigt forbrug af underlag. Disse resultater er i tæt overensstemmelse med de meget lavere (med 50�70%) niveauer af malat, pyruvat og succinat, der er blevet observeret efter en 1-times puls af [U-13C6]glukose i Keap1-KO sammenlignet med WT MEF celler [24]. I Nrf2-KO neuroner er kun pyruvat i stand til at øge ??m, hvorimod malat og methylsuccinat forårsager mild depolarisering. Virkningen af Nrf2 på mitokondriel substratproduktion synes at være den vigtigste mekanisme, hvorved Nrf2 påvirker mitokondriel funktion. Det mitokondrielle NADH-redoxindeks (balancen mellem forbrug af NADH ved kompleks I og produktion af NADPH i TCA-cyklussen) er signifikant lavere i Nrf2-KO-celler sammenlignet med deres WT-modstykker, og desuden er hastighederne for regenerering af puljerne af NADH og FADH2 efter inhibering af kompleks IV (ved brug af NaCN) er langsommere i mutantcellerne.
I mitokondrier isoleret fra murin hjerne og lever øger tilskud af substrater for kompleks I eller for kompleks II iltforbruget kraftigere, når Nrf2 aktiveres og mindre effektivt, når Nrf2 er forstyrret [35]. Således inducerer malat et højere iltforbrug i Keap1-KD sammenlignet med WT, men dets virkning er svagere i Nrf2-KO mitokondrier. Tilsvarende aktiverer succinat i nærvær af rotenon (når kompleks I hæmmes) iltforbruget i højere grad i Keap1-KD sammenlignet med WT, hvorimod responsen i Nrf2-KO mitokondrier er formindsket. Derudover er Nrf2-KO primære neuronale kulturer og mus mere følsomme over for toksiciteten af de komplekse II-hæmmere 3-nitropropionsyre og malonat, hvorimod intrastriatal transplantation af Nrf2-overudtrykkende astrocytter er beskyttende [48], [49]. Tilsvarende er Nrf2-KO-mus mere følsomme over for, hvorimod genetisk eller farmakologisk aktivering af Nrf2 har beskyttende virkninger mod neurotoksicitet forårsaget af kompleks I-hæmmeren 1-methyl-4-phenylpyridinium-ion i 1-methyl-4-phenyl-1,2,3,6, 49-tetrahydropyridin dyremodel af Parkinsons sygdom [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [XNUMX].
Respirationskontrolforholdet (RCR), forholdet mellem tilstand 3 (ADP-stimuleret) og tilstand 4 respiration (ingen ADP til stede), er nedsat i fravær af Nrf2, men RCR er ens mellem Keap1-KD og WT mitokondrier [35 ]. Da RCR er en indikation af graden af kobling af mitokondriel respiratorisk kædeaktivitet til oxidativ fosforylering, indikerer dette fund, at den højere respirationshastighed i Keap1-KD mitokondrier ikke skyldes afkobling af oxidativ fosforylering. Det tyder endvidere på, at oxidativ phosphorylering er mere effektiv, når Nrf2 er aktiveret. Den højere respirationshastighed i Keap1-KD mitokondrier er i overensstemmelse med de højere niveauer af mitokondriel ROS-produktion [38], da højere respirationshastigheder kan føre til øget elektronlækage. Men under forhold med oxidativ stress modvirkes den øgede ROS-produktion af den Nrf2-afhængige transkriptionelle opregulering af afkoblingsprotein 3 (UCP3), som øger protonkonduktansen af den mitokondrielle indre membran og som følge heraf nedsætter produktionen af superoxid [62]. For ganske nylig blev det vist, at lipidperoxidationsproduktet 4-hydroxy-2-nonenal medierer den Nrf2-afhængige opregulering af UCP3 i kardiomyocytter; dette kan være særligt vigtigt for beskyttelse under forhold med oxidativ stress, såsom dem under iskæmi-reperfusion [63].
Nrf2 påvirker effektiviteten af oxidativ phosphorylering og syntesen af ATP
I overensstemmelse med virkningen af Nrf2 på respiration, i hjerne- og levermitokondrier, resulterer Nrf2-mangel i en nedsat effektivitet af oxidativ fosforylering (som estimeret ved forholdet mellem ADP og oxygen, som forbruges til ATP-syntese), hvorimod Nrf2-aktivering (Keap1) -KD) har den modsatte effekt [35]. Sammenlignet med WT er ATP-niveauerne signifikant højere i celler med konstitutiv opregulering af Nrf2 og lavere, når Nrf2 er slået ned [64] eller forstyrret [35]. Desuden har brugen af inhibitorer af oxidativ phosphorylering (oligomycin) eller glykolyse (iodoeddikesyre) afsløret, at Nrf2 ændrer måden, hvorpå celler producerer ATP. I WT-neuroner forårsager oligomycin således et fuldstændigt fald i ATP, og iodeddikesyre har ingen yderligere effekt. Bemærkelsesværdigt, i Nrf2-KO-celler øger oligomycin ATP-niveauerne, som derefter langsomt, men fuldstændigt, udtømmes af iodeddikesyre, hvilket indikerer, at i fravær af Nrf2 er glykolyse og ikke oxidativ phosphorylering hovedkilden til ATP-produktion. Interessant nok, på trods af den øgede effektivitet af oxidativ phosphorylering i Keap1-KD-celler, resulterer tilføjelse af oligomycin i et ~80% fald i ATP-niveauer, og iodeddikesyre forårsager et yderligere ~20% fald. Således reducerer enten Nrf2-mangel eller dens konstitutive aktivering bidraget fra oxidativ phosphorylering og øger bidraget af glykolyse til syntesen af ATP. Denne effekt er især udtalt, når Nrf2 er fraværende og er i overensstemmelse med ??m'ens afhængighed af tilstedeværelsen af glukose i mediet [35] og de øgede niveauer af glykolytiske mellemprodukter (G-6-P, F-6-P , dihydroxyacetonephosphat, pyruvat og laktat) efter knockdown af Nrf2 [24].
Stigningen i ATP-niveauer efter inhibering af F1F0-ATPasen med oligomycin indikerer, at i fravær af Nrf2 fungerer F1F0-ATPasen som en ATPase og ikke en ATP-syntase, dvs. den fungerer omvendt. En sådan vending i aktivitet afspejler højst sandsynligt behovet for at pumpe protoner hen over den indre mitokondriemembran i et forsøg på at opretholde ??m, hvilket er afgørende for den funktionelle integritet af denne organel. Reverseringen af funktionen af F1F0-ATPase er også bevist af den observerede mitokondrielle depolarisering ved oligomycinadministration til Nrf2-KO-celler, hvilket er i skarp kontrast til den hyperpolarisering, der forekommer i deres WT- eller Keap1-deficiente modstykker [35]. Samlet set ser det ud til, at under forhold med Nrf2-mangel produceres ATP primært i glykolyse, og denne ATP bruges derefter delvist af F1F0-ATPasen til at opretholde ??m.
Nrf2 Forbedrer mitokondriel fedtsyreoxidation
Virkningen af Nrf2-mangel på ??m er især udtalt, når celler inkuberes i medium uden glucose, og ??m er ~50% lavere i Nrf2-KO sammenlignet med WT-celler [35]. Under forhold med glukosemangel er mitokondriel fedtsyreoxidation (FAO) en vigtig leverandør af substrater til respiration og oxidativ fosforylering, hvilket tyder på, at Nrf2 kan påvirke FAO. Faktisk er effektiviteten af FAO for både den langkædede (C16:0) mættede fedtsyre palmitinsyre og den kortkædede (C6:0) hexansyre højere i Keap1-KO MEF'er og isolerede hjerte- og levermitokondrier end i deres WT-modstykker, hvorimod det er lavere i Nrf2-KO-celler og mitokondrier [65]. Disse virkninger er også yderst relevante for mennesker: faktisk er metaboliske ændringer, der indikerer bedre integration af FAO med aktiviteten af TCA-cyklussen, blevet rapporteret at forekomme i humane interventionsstudier med diæter rig på glucoraphanin, forløberen for den klassiske Nrf2-aktivator sulforaphane [ 66].
Under det første trin af mitokondriel FAO forlader pro-R-brinten af β-carbonet som et hydrid, der reducerer FAD-cofaktoren til FADH2, som igen overfører elektroner til ubiquinon (UbQ) i respirationskæden, hvilket i sidste ende bidrager til ATP-produktion . Mens stimulering af FAO med palmitoylcarnitin i fravær af glucose forårsager den forventede stigning i ATP-niveauerne i WT- og Keap1-KO-celler, hvor ATP-stigningen er hurtigere i Keap1-KO-celler, producerer den identiske behandling ingen ATP-ændringer i Nrf2-KO MEF'er [65]. Dette eksperiment viser, at i fravær af Nrf2 undertrykkes FAO, og desuden implicerer det undertrykkelse af FAO som en af årsagerne til de lavere ATP-niveauer under betingelser med Nrf2-mangel [35], [64].
Navnlig har humane 293 T-celler, hvor Nrf2 er blevet dæmpet, en lavere ekspression af CPT1 og CPT2[67], to isoformer af carnitin palmitoyltransferase (CPT), det hastighedsbegrænsende enzym i mitokondriel FAO. I overensstemmelse hermed er mRNA-niveauerne af Cpt1 lavere i leveren af Nrf2-KO sammenlignet med WT-mus [68]. CPT katalyserer overførslen af acylgruppen i en langkædet fedtsyreacyl-CoA fra coenzym A til l-carnitin og tillader således import af acylcarnitin fra cytoplasmaet ind i mitokondrierne. Selvom dette ikke er blevet undersøgt til dato, er det muligt, at ud over de transkriptionelle virkninger på CPT1-ekspression, kan Nrf2 også påvirke funktionen af dette enzym ved at kontrollere niveauerne af dets vigtigste allosteriske inhibitor, malonyl-CoA. Dette skyldes, at Nrf2 ved en mekanisme, der i øjeblikket er uklar, negativt regulerer ekspressionen af stearoyl CoA desaturase (SCD) [69] og citratlyase (CL) [69], [70]. Mærkeligt nok fører knockout eller inhibering af SCD til øget phosphorylering og aktivering af AMP-aktiveret proteinkinase (AMPK) [71], [72], [73], og det kan spekuleres i, at i fravær af Nrf2, SCD-niveauerne vil øge, til gengæld sænke AMPK-aktiviteten. Dette kan yderligere forstærkes af de reducerede proteinniveauer af AMPK, der er blevet observeret i leveren af Nrf2-KO-mus [68], et fund, der er i tæt overensstemmelse med de øgede AMPK-niveauer, som er blevet rapporteret i leveren af Keap1-KD mus [74]. En konsekvens af den nedsatte AMPK-aktivitet er lindring af dens hæmmende phosphorylering (ved Ser79) af acetyl-CoA-carboxylase (ACC) [75], som kunne opreguleres yderligere transkriptionelt i fravær af Nrf2, fordi den nedreguleres af Nrf2-aktivering [70] ]. Den høje ACC-aktivitet i kombination med den opregulerede CL-ekspression, der vil øge produktionen af acetyl-CoA, substratet for ACC, kan i sidste ende øge niveauerne af ACC-produktet, malonyl-CoA. De høje niveauer af malonyl-CoA vil hæmme CPT og derved mindske transporten af fedtsyrer ind i mitokondrierne. Endelig regulerer Nrf2 positivt ekspressionen af CD36 [76], en translokase, der importerer fedtsyrer over plasma og mitokondriemembraner. Således er en mekanisme, hvorved Nrf2 kan påvirke effektiviteten af mitokondriel FAO, ved at regulere importen af langkædede fedtsyrer til mitokondrierne.
Ud over direkte transkriptionel regulering kan Nrf2 også ændre effektiviteten af mitokondriel FAO ved dets virkninger på den cellulære redoxmetabolisme. Dette kan være særligt relevant, når Nrf2-aktivitet er lav eller fraværende, tilstande, der skifter den cellulære redoxstatus mod den oxiderede tilstand. Faktisk er flere FAO-enzymer blevet identificeret som følsomme over for redoxændringer. Et sådant enzym er meget langkædet acyl-CoA-dehydrogenase (VLCAD), som bidrager med mere end 80 % til palmitoyl-CoA-dehydrogeneringsaktiviteten i humant væv [77]. Interessant nok, Hurd et al. [78] har vist, at VLCAD indeholder cysteinrester, der signifikant ændrer deres redoxtilstand ved eksponering af isolerede rottehjertemitokondrier for H2O2. Derudover forbedrer S-nitrosylering af murint hepatisk VLCAD ved Cys238 den katalytiske effektivitet af enzymet [79], og det er sandsynligt, at oxidation af det samme cystein kan have den modsatte effekt, hvilket i sidste ende sænker effektiviteten af mitokondriel FAO. Det er derfor muligt, at selvom ekspressionsniveauerne af VLCAD ikke er signifikant forskellige i WT, Nrf2-KO eller Keap1-KO MEF'er [65], kan enzymaktiviteten af VLCAD være lavere i fravær af Nrf2 på grund af de højere niveauer af ROS.
Baseret på alle disse resultater kan det foreslås, at (fig. 3): i fravær af Nrf2 er NADPH-niveauerne lavere på grund af nedsat ekspression af ME1, IDH1, G6PD og PGD. Niveauerne af reduceret glutathion er også lavere på grund af nedsat ekspression af enzymer, der deltager i dets biosyntese og regenerering, og de lavere niveauer af NADPH, der er nødvendige for omdannelsen af den oxiderede til den reducerede form af glutathion. Den lave ekspression af ME1 vil mindske puljen af pyruvat, der kommer ind i mitokondrierne, hvor glykolyse bliver den vigtigste kilde til pyruvat. Genereringen af NADH er langsommere, hvilket fører til nedsat aktivitet af kompleks I og øget mitokondriel ROS-produktion. Reduktionen af FAD til FADH2 er også langsommere, i det mindste delvist på grund af mindre effektiv fedtsyreoxidation, hvilket kompromitterer elektronstrømmen fra FADH2 til UbQ og ind i kompleks III. Da UbQH2 er en aktivator af succinatdehydrogenase [80], kan nedsættelse af dets dannelse sænke enzymaktiviteten af succinatdehydrogenase. De øgede niveauer af superoxid og hydrogenperoxid kan hæmme kompleks II-aktivitet yderligere [81]. Den lavere effektivitet af fedtsyreoxidation bidrager til den reducerede substrattilgængelighed for mitokondriel respiration og ATP-produktion ved oxidativ fosforylering. Som en kompenserende mekanisme forstærkes glykolysen. ATP-syntase fungerer omvendt som en ATPase i et forsøg på at opretholde ??m.
Nrf2 og mitokondriel biogenese
Det er blevet rapporteret, at sammenlignet med WT har leveren af Nrf2-KO mus et lavere mitokondrieindhold (som bestemt af forholdet mellem mitokondrie og nukleært DNA); dette reduceres yderligere med en 24-timers faste i både WT- og Nrf2-KO-mus; i modsætning hertil, selvom det ikke er forskelligt fra WT under normale fodringsforhold, påvirkes mitokondrieindholdet i mus med høj Nrf2-aktivitet ikke af faste [82]. Interessant nok fremmer tilskud med Nrf2-aktivatoren (R)-a-liponsyre [83], [84], [85] mitokondriel biogenese i 3T3-L1-adipocytter [86]. To klasser af nukleare transkriptionelle regulatorer spiller kritiske roller i mitokondriel biogenese. Den første klasse er transkriptionsfaktorer, såsom nukleære respiratoriske faktorer11 og 2, som styrer ekspressionen af gener, der koder for underenheder af de fem respiratoriske komplekser, mitokondrielle translationskomponenter og hæmbiosyntetiske enzymer, der er lokaliseret til mitokondriematrixen [88]. Piantadosi et al. [89] har vist, at den Nrf2-afhængige transkriptionelle opregulering af nuklear respiratorisk faktor 1 fremmer mitokondriel biogenese og beskytter mod cytotoksiciteten af det kardiotoksiske antracyklin kemoterapeutiske middel doxorubicin. I modsætning hertil har Zhang et al. [82] har rapporteret, at genetisk aktivering af Nrf2 ikke påvirker den basale mRNA-ekspression af nuklear respiratorisk faktor 1 i den murine lever.
Den anden klasse af nukleare transkriptionelle regulatorer med kritiske funktioner i mitokondriel biogenese er transkriptionelle coaktivatorer, såsom peroxisomproliferator-aktiveret receptor ? coaktivatorer (PGC)1? og 1?, som interagerer med transkriptionsfaktorer, det basale transkriptionelle og RNA-splejsningsmaskineri og histonmodificerende enzymer [88], [90], [91]. Ekspressionen af PGC1-familien af coaktivatorer er påvirket af adskillige miljøsignaler. Behandling af humane fibroblaster med Nrf2-aktivatoren sulforaphane forårsager en stigning i mitokondriel masse og induktion af PGC1? og PGC1? [92], selvom den potentielle afhængighed af Nrf2 ikke blev undersøgt i denne undersøgelse. Diabetiske mus, hvor Nrf2 enten aktiveres af Keap1-genets hypomorfiske knockdown (db/db:Keap1flox/?:Nrf2+/+) eller forstyrrede (db/db:Keap1flox/?:Nrf2?/?) har lavere lever-PGC1? ekspressionsniveauer end kontroldyr (db/db:Keap1flox/+:Nrf2+/+) [93]. Ingen forskelle i mRNA-niveauerne for PGC1? ses i leveren fra ikke-diabetiske mus, der er enten WT eller Nrf2-KO, hvorimod disse niveauer er lavere i Nrf2-overudtrykkende (Keap1-KD og leverspecifikke Keap1-KO) dyr [82]. Især øger en 24-timers faste niveauerne af PGC1? mRNA i leveren af mus af alle genotyper, men stigningen er signifikant større i leveren af Nrf2-KO sammenlignet med WT eller Nrf2-overudtrykkende mus. Sammenlignet med WT viser Nrf2-KO-mus, der oplever septisk infektion eller akut lungeskade på grund af infektion, svækket transkriptionel opregulering af nuklear respiratorisk faktor 1 og PGC1? [94], [95]. Sammen antyder disse observationer, at Nrf2's rolle i at opretholde niveauerne af både nuklear respiratorisk faktor 1 og PGC1? er kompleks og bliver mest fremtrædende under stresstilstande.
Ud over ekspression af gener, der koder for mitokondrielle proteiner, kræver mitokondriel biogenese syntese af nukleotider. Genetisk aktivering af Nrf2 øger purinbiosyntesen ved at opregulere pentosephosphatvejen og metabolismen af folat og glutamin, især i hurtigt prolifererende celler (fig. 2) [24]. Analyse af transkriptomet af mutant Drosophila, der mangler den mitokondrielle serin/threoninproteinkinase PTEN-induceret formodet kinase 1 (PINK1) har vist, at mitokondriel dysfunktion fører til transkriptionel opregulering af gener, der påvirker nukleotidmetabolismen [96], hvilket tyder på, at det forbedrede nukleotidbiosyntese-nukleotid repræsenterer en mekanisme til beskyttelse mod de neurotoksiske konsekvenser af PINK1-mangel. Nrf2 regulerer ekspressionen af phosphoribosyl pyrophosphat amidotransferase (PPAT), som katalyserer indgangen i den de novo purin nukleotid biosyntesevej, og mitokondriel methylentetrahydrofolat dehydrogenase 2 (MTHFD2) (fig. 2). Sidstnævnte er et bifunktionelt enzym med dehydrogenase- og cyclohydrolaseaktiviteter, der er afgørende for at tilvejebringe både glycin og formiat som kilder til en-carbon-enheder til purinbiosyntese i hurtigt voksende celler [97]. Det er derfor sandsynligt, at Nrf2-aktivering kan være beskyttende og kan vende mitokondriel dysfunktion i PINK1-mangel. Faktisk genopretter farmakologisk aktivering af Nrf2 af sulforaphane eller triterpenoidet RTA-408 µm og beskytter PINK1-mangelfulde celler mod dopamintoksicitet [98]. Selvom de underliggende mekanismer synes at være komplekse, tilsammen indikerer disse resultater, at Nrf2-aktivitet kan påvirke mitokondriel biogenese ved at påvirke ekspressionsniveauerne af kritiske transkriptionsfaktorer og coaktivatorer, såvel som ved at øge nukleotidbiosyntesen.
Nrf2 og mitokondriel integritet
Selvom direkte beviser ikke altid er tilgængelige, er der stærke indikationer på, at Nrf2 er vigtig for mitokondriel integritet, især under forhold med oxidativ stress. Mitokondrier isoleret fra hjernen og leveren hos rotter, der havde fået en enkelt dosis af Nrf2-aktivatoren sulforaphane, er modstandsdygtige over for åbning af mitokondriel permeabilitetsovergangspore (mPTP) forårsaget af oxidanten tert-butylhydroperoxid [99], [100]. mPTP, et kompleks, der gør det muligt for den mitokondriske indre membran at blive permeabel for molekyler med masser op til 1500 Da, blev for nylig identificeret til at være dannet fra dimerer af F0F1-ATP-syntasen [101]. Den sulforaphane-medierede resistens over for mPTP-åbning korrelerer med øget antioxidantforsvar, og niveauerne af mitokondriel GSH, glutathionperoxidase 1, æblesyreenzym 3 og thioredoxin 2 er alle opreguleret i mitokondrielle fraktioner isoleret fra sulforaphan-behandlede dyr [100].
Mitokondriel proteinskade og svækkelse af respiration forårsaget af det elektrofile lipidperoxidationsprodukt 4-hydroxy-2-nonenal svækkes i mitokondrier isoleret fra hjernebarken hos sulforaphane-behandlede mus [102]. I nyreepitelceller fra rotter og i nyrer er sulforaphan beskyttende mod cisplatin- og gentamicin-induceret toksicitet og tab af ??m[103], [104]. Beskyttelse mod et panel af oxidanter (superoxid, hydrogenperoxid, peroxynitrit) og elektrofiler (4-hydroxy-2-nonenal og acrolein) og en stigning i mitokondrielle antioxidantforsvar er også blevet observeret ved behandling af glatte muskelceller fra rotteaorta med sulforaphane [105 ]. I en model af kontrast-induceret akut nyreskade, blev iskæmisk prækonditionering af lemmer for nylig vist at have beskyttende virkninger, herunder hæmning af åbningen af mPTP og mitokondriel hævelse, ved aktivering af Nrf2 som følge af hæmningen af GSK3? [106].
Mitofagi, den proces, hvorved dysfunktionelle mitokondrier selektivt opsluges af autophagosomer og leveres til lysosomer for at blive nedbrudt og genanvendt af cellen, er afgørende for mitokondriel homeostase [107], [108]. Mens der ikke er etableret en årsagssammenhæng mellem Nrf2 og mitofagi, er der bevis for, at transkriptionsfaktoren kan være vigtig i mitokondriel kvalitetskontrol ved at spille en rolle i mitofagi. Dette kan være særligt fremtrædende under forhold med oxidativ stress. I en model af sepsis er stigningerne i niveauerne af autophagosommarkøren MAP1 let kæde 3-II (LC3-II) og cargoproteinet p62 24 timer efter infektion således undertrykt i Nrf2-KO sammenlignet med WT-mus [109] . En lille molekyle inducer af mitofagi (kaldet p62-medieret mitophagy inducer, PMI) blev for nylig opdaget; denne 1,4-diphenyl-1,2,3-triazolforbindelse blev oprindeligt designet som en Nrf2-aktivator, der forstyrrer interaktionen af transkriptionsfaktoren med Keap1 [110]. I lighed med celler, hvor Nrf2 er genetisk opreguleret (Keap1-KD eller Keap1-KO), har celler udsat for PMI højere hvilende ??m. Det er vigtigt, at stigningen i mitokondriel LC3-lokalisering, der observeres efter PMI-behandling af WT-celler, ikke forekommer i Nrf2-KO-celler, hvilket tyder på involvering af Nrf2.
Sidst har ultrastrukturel analyse af leversektioner afsløret tilstedeværelsen af hævede mitokondrier med reduceret crista og forstyrrede membraner i hepatocytter af Nrf2-KO, men ikke WT, mus, der var blevet fodret med en kost med højt fedtindhold i 24 uger; især viser disse lever tydelige tegn på oxidativt stress og inflammation [68]. Det kan konkluderes, at Nrf2 har en afgørende rolle i at opretholde mitokondriel integritet under forhold med oxidativt og inflammatorisk stress.
Sulforaphane og dens virkninger på kræft, dødelighed, aldring, hjerne og adfærd, hjertesygdomme og mere
Isothiocyanater er nogle af de vigtigste planteforbindelser, du kan få i din kost. I denne video gør jeg det mest omfattende tilfælde til dem, der nogensinde er blevet lavet. Kort opmærksomhed span? Spring til dit yndlingsemne ved at klikke på et af nedenstående tidspunkter. Fuld tidslinje nedenfor.
Nøglesektioner:
00: 01: 14 - Kræft og dødelighed
00: 19: 04 - Aging
00: 26: 30 - Hjerne og adfærd
00: 38: 06 - Final recap
00: 40: 27 - Dosis
Fuld tidslinje:
00: 00: 34 - Introduktion af sulforaphane, et vigtigt fokus i videoen.
00: 01: 14 - Cruciferous vegetabilsk forbrug og reduktion af allårsag dødelighed.
00: 02: 12 - Prostatacancerrisiko.
00: 02: 23 - Kræftrisiko.
00: 02: 34 - Lungekræft i rygere risiko.
00: 02: 48 - Brystkræftrisiko.
00: 03: 13 - Hypotetisk: Hvad hvis du allerede har kræft? (Interventionel)
00: 03: 35 - Plausibel mekanisme, der driver kræft- og dødelighedsassosierende data.
00: 04: 38 - Sulforaphane og kræft.
00: 05: 32 - Dyrebevis, der viser stærk virkning af broccoli-spireekstrakt på udviklingen af blæretumor hos rotter.
00: 06: 06 - Virkning af direkte tilsætning af sulforaphan hos patienter med prostatacancer.
00: 07: 09 - Bioakkumulering af isothiocyanatmetabolitter i egentligt brystvæv.
00: 08: 32 - Inhibering af brystcancerstamceller.
00: 08: 53 - Historie lektion: brassicas blev etableret som at have sundhedsegenskaber selv i det gamle Rom.
00: 09: 16 - Sulforaphans evne til at øge kræftfremkaldende udskillelse (benzen, acrolein).
00: 09: 51 - NRF2 som en genetisk switch via antioxidant responselementer.
00: 10: 10 - Hvordan NRF2 aktivering øger kræftfremkaldende udskillelse via glutathion-S-konjugater.
00: 10: 34 - Spiraler øger glutathion-S-transferase og reducerer DNA-beskadigelse.
00: 11: 20 - Broccoli-spire drikker øger benzenudskillelsen med 61%.
00: 13: 31 - Broccoli spirehomogenat øger antioxidant enzymer i den øvre luftvej.
00: 15: 45 - Cruciferous vegetabilsk forbrug og dødelighed i hjertesygdomme.
00: 16: 55 - Broccoli spire pulver forbedrer blodlipider og overordnet hjertesygdomsrisiko hos type 2 diabetikere.
00: 19: 04 - Begyndelse af aldringssektionen.
00: 19: 21 - Sulforaphane beriget kost forbedrer levetiden for biller fra 15 til 30% (under visse forhold).
00: 20: 34 - Betydningen af lav inflammation for lang levetid.
00: 22: 05 - Cruciferous grøntsager og broccoli spire pulver synes at reducere et bredt udvalg af inflammatoriske markører hos mennesker.
00: 24: 14 - Musestudier tyder på, at sulforaphan kan forbedre adaptiv immunfunktion i alderdommen.
00: 25: 18 - Sulforaphane forbedrede hårvækst i en musmodel af skaldethed. Billede på 00: 26: 10.
00: 26: 30 - Begyndelse af hjerne- og adfærdssektionen.
00: 27: 18 - Effekt af broccoli-spireekstrakt på autisme.
00: 27: 48 - Virkning af glucoraphanin på skizofreni.
00: 28: 17 - Start af depression diskussion (sandsynlig mekanisme og undersøgelser).
00: 31: 21 - Mus undersøgelse ved hjælp af 10 forskellige modeller af stress-induceret depression viser sulforaphan på samme måde som fluoxetin (prozac).
00: 32: 00 - Undersøgelse viser direkte indtagelse af glucoraphanin hos mus er tilsvarende effektiv til forebyggelse af depression fra social nederlagsspændingsmodel.
00: 33: 01 - Begyndelsen af neurodegenerationsafsnittet.
00: 33: 30 - Sulforaphane og Alzheimers sygdom.
00: 33: 44 - Sulforaphane og Parkinsons sygdom.
00: 33: 51 - Sulforaphane og Hungtington's sygdom.
00: 35: 55 - Sulforaphane og neuronal plasticitet.
00: 36: 32 - Sulforaphane forbedrer læring som model af type II diabetes hos mus.
00: 37: 19 - Sulforaphane og duchenne muskeldystrofi.
00: 37: 44 - Myostatininhibering i muskel-satellitceller (in vitro).
00: 38: 06 - Late video recap: dødelighed og kræft, DNA-beskadigelse, oxidativ stress og betændelse, udslip af benzen, kardiovaskulær sygdom, type II diabetes, hjernens virkninger (depression, autisme, skizofreni, neurodegeneration), NRF2-pathway.
00: 40: 27 - Tanker om at finde ud af en dosis broccolispirer eller sulforaphane.
00: 41: 01 - Anecdoter på spiring hjemme.
00: 43: 14 - På tilberedningstemperaturer og sulforafanaktivitet.
00: 43: 45 - Gut-bakteriekonvertering af sulforaphan fra glucoraphanin.
00: 44: 24 - Tilskud fungerer bedre, når de kombineres med aktiv myrosinase fra grøntsager.
00: 44: 56 - Madlavningsteknikker og cruciferous grøntsager.
00: 46: 06 - Isothiocyanater som goitrogener.
Nrf2 er en transkriptionsfaktor, som spiller en vigtig rolle i den menneskelige krops cellulære antioxidantforsvar. Det antioxidant-responsive element, eller ARE, er en reguleringsmekanisme af gener. Mange forskningsundersøgelser har vist, at Nrf2, eller NF-E2-relateret faktor 2, regulerer en lang række ARE-drevne gener gennem flere typer celler. Nrf2 viste sig også at spille en væsentlig rolle i cellulær beskyttelse og anti-carcinogenicitet, hvilket viser, at Nrf2 kan være en effektiv behandling i behandlingen af neurodegenerative sygdomme og kræftformer, der menes at være forårsaget af oxidativt stress. Dr. Alex Jimenez DC, CCST Insight
Afsluttende bemærkninger
Selvom mange spørgsmål stadig er åbne, indikerer det tilgængelige eksperimentelle bevis klart, at Nrf2 er en vigtig aktør i opretholdelsen af mitokondriel homeostase og strukturel integritet. Denne rolle bliver særligt kritisk under forhold med oxidativ, elektrofil og inflammatorisk stress, når evnen til at opregulere Nrf2-medierede cytobeskyttende responser påvirker cellens og organismens generelle sundhed og overlevelse. Rollen af Nrf2 i mitokondriel funktion repræsenterer et andet lag af de brede cytobeskyttende mekanismer orkestreret af denne transkriptionsfaktor. Da mange menneskelige patologiske tilstande har oxidativt stress, inflammation og mitokondriel dysfunktion som væsentlige komponenter i deres patogenese, lover farmakologisk aktivering af Nrf2 et løfte om sygdomsforebyggelse og behandling. Omfattende forståelse af de præcise mekanismer, hvorved Nrf2 påvirker mitokondriel funktion, er afgørende for rationelt design af fremtidige kliniske forsøg og kan tilbyde nye biomarkører til overvågning af terapeutisk effekt.
Formålet med artiklen ovenfor var at diskutere såvel som at demonstrere den nye rolle af Nrf2 i mitokondriel funktion. Nrf2, eller nuklear faktor erythroid 2-relateret faktor, er en ny regulator af cellulær resistens over for oxidanter, som kan bidrage til oxidativt stress, som påvirker cellulær funktion og fører til udvikling af toksicitet, kronisk sygdom og endda kræft. Mens produktionen af oxidanter i den menneskelige krop kan tjene forskellige formål, herunder celledeling, inflammation, immunfunktion, autofagi og stressrespons, er det vigtigt at kontrollere deres overproduktion for at forhindre sundhedsproblemer. Omfanget af vores information er begrænset til kiropraktik og rygsøjlesundhedsproblemer. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Rygsmerte er en af de mest udbredte årsager til handicap og ubesvarede arbejdsdage over hele verden. Rygsmerter tilskrives den næst hyppigste årsag til lægekontorbesøg, der kun er større end infektioner i de øvre luftveje. Ca. 80 procent af befolkningen vil opleve rygsmerter mindst én gang i hele deres liv. Rygsøjlen er en kompleks struktur, der består af knogler, led, ledbånd og muskler, blandt andet blødt væv. På grund af dette kan kvæstelser og / eller forværrede forhold, såsom herniated diske, kan i sidste ende føre til symptomer på rygsmerter. Sportsskader eller bilulykkesskader er ofte den hyppigste årsag til rygsmerter, men nogle gange kan de enkleste bevægelser have smertefulde resultater. Heldigvis kan alternative behandlingsmuligheder, såsom kiropraktisk pleje, hjælpe med at lindre rygsmerter gennem brug af spinaljusteringer og manuelle manipulationer, hvilket i sidste ende forbedrer smertelindring. �
Nrf2 understøtter aktiveringen af en gruppe antioxidanter og afgiftende enzymer og gener, som beskytter den menneskelige krop mod virkningerne af sundhedsproblemer forbundet med øgede niveauer af oxidativt stress, såsom Alzheimers sygdom. En række naturlige stoffer har vist sig at aktivere Nrf2-vejen, som kan hjælpe med at håndtere symptomerne på neurodegenerative sygdomme. Formålet med artiklen nedenfor er at diskutere den centrale rolle af Nrf2 forårsaget af kronisk inflammation.
Abstrakt
Betændelse er det mest almindelige træk ved mange kroniske sygdomme og komplikationer, mens det spiller en afgørende rolle i carcinogenese. Adskillige undersøgelser har vist, at Nrf2 bidrager til den antiinflammatoriske proces ved at orkestrere rekruttering af inflammatoriske celler og regulere genekspression gennem antioxidantresponselementet (ARE). Keap1 (Kelch-lignende ECH-associeret protein)/Nrf2 (NF-E2 p45-relateret faktor 2)/ARE signalvejen regulerer hovedsageligt anti-inflammatorisk genekspression og hæmmer progressionen af inflammation. Derfor er identifikationen af nye Nrf2-afhængige antiinflammatoriske fytokemikalier blevet et nøglepunkt i lægemiddelopdagelsen. I denne gennemgang diskuterer vi medlemmerne af Keap1/Nrf2/ARE-signalvejen og dens nedstrømsgener, virkningerne af denne vej på dyremodeller af inflammatoriske sygdomme og krydstale med NF-?B-vejen. Derudover diskuterer vi også om reguleringen af NLRP3-inflammasom af Nrf2. Udover dette opsummerer vi det aktuelle scenarie for udvikling af antiinflammatoriske fytokemikalier og andre, der formidler Nrf2/ARE-signalvejen.
Inflammation er en kompleks proces, der opstår, når væv er inficeret eller skadet af skadelige stimuli såsom patogener, skader eller irriterende stoffer. Immunceller, blodkar og molekylære mediatorer er involveret i dette beskyttende respons [1]. Inflammation er også et patologisk fænomen forbundet med en række sygdomstilstande, der hovedsageligt induceres af fysiske, kemiske, biologiske og psykologiske faktorer. Formålet med inflammation er at begrænse og eliminere årsagerne til cellulær skade, rense og/eller absorbere nekrotiske celler og væv og igangsætte vævsreparation. Der skelnes mellem to forskellige former for betændelse: akut og kronisk. Akut inflammation er selvbegrænsende og gavnlig for værten, men langvarig kronisk inflammation er et fællestræk ved mange kroniske sygdomme og komplikationer. Direkte infiltration af mange mononukleære immunceller såsom monocytter, makrofager, lymfocytter og plasmaceller, såvel som produktionen af inflammatoriske cytokiner, fører til kronisk inflammation. Det er anerkendt, at kronisk inflammation spiller en afgørende rolle i carcinogenese [2]. Generelt interagerer både pro- og antiinflammatoriske signalveje i den normale inflammatoriske proces.
I den patologiske inflammatoriske proces aktiveres først mastceller, monocytter, makrofager, lymfocytter og andre immunceller. Derefter rekrutteres cellerne til skadestedet, hvilket resulterer i dannelsen af reaktive oxygenarter (ROS), der beskadiger makromolekyler inklusive DNA. Samtidig producerer disse inflammatoriske celler også store mængder af inflammatoriske mediatorer såsom cytokiner, kemokiner og prostaglandiner. Disse mediatorer rekrutterer yderligere makrofager til lokaliserede inflammationssteder og aktiverer direkte flere signaltransduktionskaskader og transkriptionsfaktorer forbundet med inflammation. NF-?B (nuklear faktor kappa B), MAPK (mitogen-aktiveret proteinkinase) og JAK (janus kinase)-STAT (signaltransducere og transkriptionsaktivatorer) er involveret i udviklingen af den klassiske inflammationsvej [3], [4], [5]. Tidligere undersøgelser har afsløret, at transkriptionsfaktoren Nrf2 (NF-E2 p45-relateret faktor 2) regulerer ekspressionen af fase II afgiftende enzymer, herunder NADPH, NAD(P)H quinonoxidoreduktase 1, glutathionperoxidase, ferritin, hæmoxygenase-1 (HO). -1), og antioxidantgener, der beskytter celler mod forskellige skader via deres antiinflammatoriske virkninger og dermed påvirker sygdomsforløbet [6], [7], [8].
I betragtning af disse bemærkelsesværdige fund har udviklingen af målrettede terapeutiske lægemidler til inflammatoriske sygdomme via signalveje tiltrukket sig stor interesse i de senere år. I denne gennemgang opsummerer vi forskning om Keap1 (Kelch-lignende ECH-associeret protein)/Nrf2 (NF-E2 p45-relateret faktor 2)/ARE (antioxidantresponselement) signalvejen i inflammation.
Struktur og regulering af Nrf2
Keap1-afhængig Nrf2-regulering
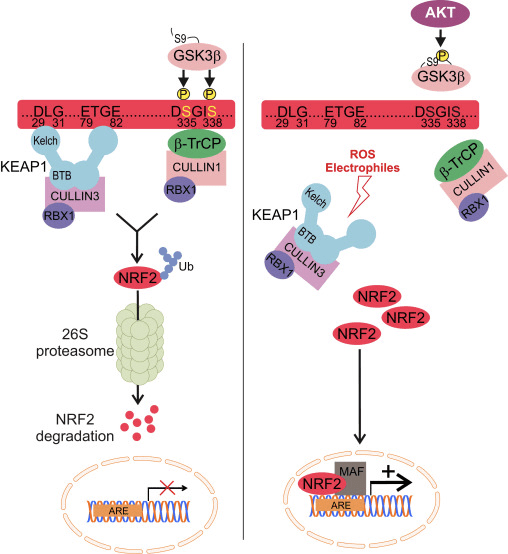
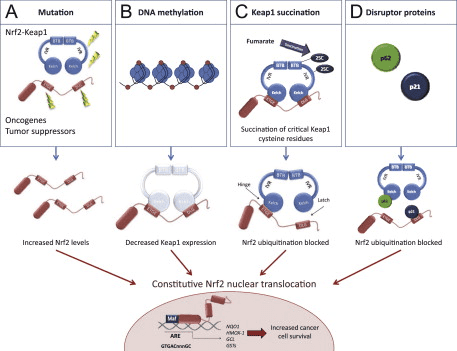
Nrf2 tilhører Cap �n� Collar (CNC) underfamilien og omfatter i syv funktionelle domæner, Neh (Nrf2-ECH homologi) 1 til Neh7 [9], [10]. Neh1 er et CNC-bZIP-domæne, der tillader Nrf2 at heterodimerisere med små muskuloaponeurotisk fibrosarkom (Maf) protein, DNA og andre transkriptionspartnere samt danne et nukleært kompleks med det ubiquitin-konjugerende enzym UbcM2 [11], [12]. Neh2 indeholder to vigtige motiver kendt som DLG og ETGE, som er essentielle for interaktionen mellem Nrf2 og dens negative regulator Keap1 [13], [14].
Keap1 er en substratadapter til cullin-baseret E3 ubiquitin-ligase, som hæmmer transkriptionsaktiviteten af Nrf2 via ubiquitinering og proteasomal nedbrydning under normale forhold [15], [16], [17]. KELCH-domænerne af Keap1-homodimeren binder til DLG- og ETGE-motiverne af Nrf2-Neh2-domænet i cytosolen, hvor ETGE fungerer som et hængsel med højere affinitet, og DLG fungerer som en latch [18]. Under oxidativ stress eller ved udsættelse for Nrf2-aktivatorer dissocierer Nrf2 fra Keap1-binding på grund af thiolmodifikationen af Keap1-cysteinrester, som i sidste ende forhindrer Nrf2-ubiquitinering og proteasomal nedbrydning [19]. Derefter translokerer Nrf2 ind i kernen, heterodimeriserer med små Maf-proteiner og transaktiverer et ARE-batteri af gener (fig. 1A). Carboxy-terminalen af Neh3 fungerer som et transaktiveringsdomæne ved at interagere med transkriptions-co-aktivatoren kendt som CHD6 (chromo-ATPase/helicase DNA-binding protein) [20]. Neh4 og Neh5 fungerer også som transaktiveringsdomæner, men binder til en anden transkriptionel co-aktivator kendt som CBP (cAMP-respons-element-bindende protein-bindende protein) [21]. Desuden interagerer Neh4 og Neh5 med den nukleare cofaktor RAC3/AIB1/SRC-3, hvilket fører til forbedret Nrf2-målrettet ARE-genekspression [22]. Neh5 har et redox-følsomt nuklear-eksportsignal, som er afgørende for reguleringen og cellulær lokalisering af Nrf2 [23].
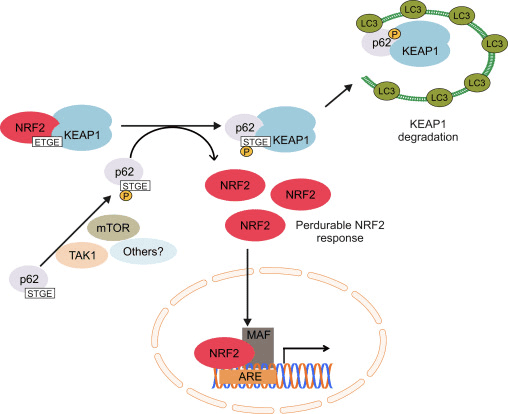
Figur 1 Keap1-afhængig og -uafhængig regulering af Nrf2. (A) Under basale forhold er Nrf2 sekvestreret med Keap1 af dets to motiver (ETGE og DLG), der fører til CUL3-medieret ubiquitinering efterfulgt af proteasomnedbrydning. Under oxidativ stress dissocierer Nrf2 fra Keap1, translokerer til kernen og aktiverer ARE-genbatteriet. (B) GSK3 phosphorylerer Nrf2, og dette letter genkendelsen af Nrf2 af a-TrCP til CUL1-medieret ubiquitinering og efterfølgende proteasomnedbrydning. (C) p62 er sekvestreret med Keap1, hvilket fører til dets autofagiske nedbrydning, frigørelsen af Nrf2 og øget Nrf2-signalering.
Keap1-uafhængig Nrf2-regulering
Nye beviser har afsløret en ny mekanisme for Nrf2-regulering, der er uafhængig af Keap1. Det serinrige Neh6-domæne af Nrf2 spiller en afgørende rolle i denne regulering ved at binde med dets to motiver (DSGIS og DSAPGS) til β-transducin-gentagelsesholdigt protein (β-TrCP) [24]. ?-TrCP er en substratreceptor for Skp1�Cul1�Rbx1/Roc1 ubiquitin-ligasekomplekset, der er rettet mod Nrf2 til ubiquitinering og proteasomal nedbrydning. Glykogensyntase-kinase-3 er et afgørende protein involveret i Keap1-uafhængig Nrf2-stabilisering og -regulering; det phosphorylerer Nrf2 i Neh6-domænet for at lette genkendelsen af Nrf2 af a-TrCP og efterfølgende proteinnedbrydning [25] (fig. 1B).
Andre Nrf2 regulatorer
En anden evidenslinje har afsløret en ikke-kanonisk vej for p62-afhængig Nrf2-aktivering, hvor p62 sekvestrerer Keap1 til autofagisk nedbrydning, der i sidste ende fører til stabilisering af Nrf2 og transaktivering af Nrf2-afhængige gener [26], [27], [ 28], [29] (fig. IC).
Akkumulerende beviser tyder på, at flere miRNA'er spiller en vigtig rolle i reguleringen af Nrf2-aktiviteten [30]. Sangokoya et al. [31] demonstrerede, at miR-144 direkte nedregulerer Nrf2-aktivitet i lymfoblast-K562-cellelinjen, primære humane erythroide progenitorceller og seglcelle-sygdomsretikulocytter. En anden interessant undersøgelse i humane brystepitelceller viste, at miR-28 hæmmer Nrf2 gennem en Keap1-uafhængig mekanisme [32]. Tilsvarende nedregulerer miRNA'er såsom miR-153, miR-27a, miR-142-5p og miR144 Nrf2-ekspression i den neuronale SH-SY5Y-cellelinje [33]. Singh et al. [34] viste, at den ektopiske ekspression af miR-93 nedsætter ekspressionen af Nrf2-regulerede gener i en 17β-østradiol (E2)-induceret rottemodel af brystkræft.
En nylig opdagelse fra vores laboratorium identificerede en endogen inhibitor af Nrf2 kendt som retinoic X receptor alpha (RXR?). RXR? er en nuklear receptor, interagerer med Neh7-domænet af Nrf2 (aminosyrerester 209�316) via dets DNA-bindende domæne (DBD), og hæmmer specifikt Nrf2-aktivitet i kernen. Desuden er andre nukleare receptorer, såsom peroxisomproliferator-aktiveret receptor-a, ERa, østrogen-relaterede receptor-a og glukokortikoidreceptorer også blevet rapporteret at være endogene hæmmere af Nrf2-aktivitet [9], [10].
Anti-inflammatorisk rolle for Nrf2/HO-1-aksen
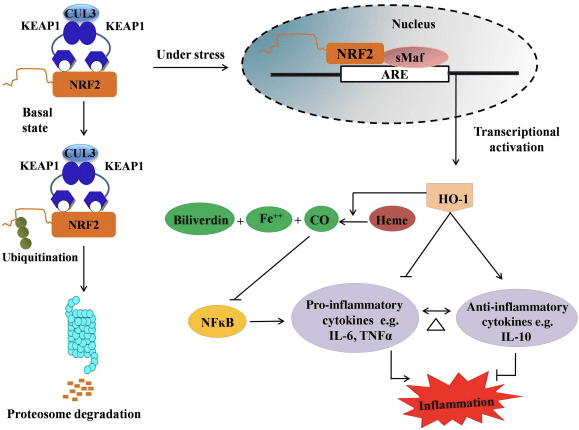
HO-1 er den inducerbare isoform og hastighedsbegrænsende enzym, der katalyserer nedbrydningen af hæm til carbonmonoxid (CO) og frit jern og biliverdin til bilirubin. Enzymatisk nedbrydning af pro-inflammatorisk fri hæm samt produktionen af anti-inflammatoriske forbindelser såsom CO og bilirubin spiller store roller i opretholdelsen af de beskyttende virkninger af HO-1 (fig. 2).
Figur 2 Oversigt over Nrf2/HO-1-vejen. Under basale forhold binder Nrf2 til sin repressor Keap1, hvilket fører til ubiquitinering efterfulgt af proteasomnedbrydning. Under oxidativ stress translokerer fri Nrf2 til kernen, hvor den dimeriserer med medlemmer af den lille Maf-familie og binder sig til ARE-gener såsom HO-1. Opreguleret HO-1 katalyserer hæmen til CO, bilirubin og frit jern. CO virker som en hæmmer af NF-?B-vejen, som fører til nedsat ekspression af pro-inflammatoriske cytokiner, mens bilirubin også virker som antioxidant. Ydermere hæmmer HO-1 direkte de proinflammatoriske cytokiner samt aktiverer de antiinflammatoriske cytokiner, hvilket fører til afbalancering af den inflammatoriske proces.
Nrf2 inducerer HO-1-genet ved at øge mRNA- og proteinekspression, og det er et af de klassiske Nrf2-regulerede gener, som er meget udbredt i talrige in vitro og in vivo undersøgelser. Adskillige undersøgelser har vist, at HO-1 og dets metabolitter har signifikante antiinflammatoriske virkninger medieret af Nrf2. Forhøjelse af HO-1-ekspression, som medieres af aktiveret Nrf2, fører til inhibering af NF?B-signalering resulterer i den reducerede tarmslimhindeskade og tight-junction-dysfunktion i levertransplantationsmodel for han-Spague-Dawley-rotter [35]. Opregulering af Nrf2-afhængig HO-1-ekspression kan beskytte muse-afledte C2C12-myoblaster mod H2O2-cytotoksicitet [36]. Nrf2-afhængig HO-1 har en indvirkning på lipopolysaccharid (LPS)-medierede inflammatoriske responser i RAW264.7- eller muse peritoneale makrofag-afledte skumcelle-makrofager. Nrf2-aktivitet desensibiliserede skumcellemakrofager fænotyper og forhindrer umådelig inflammation af makrofager, de spiller en vigtig rolle i progression af åreforkalkning [37]. Nrf2/HO-1-aksen påvirker LPS-inducerede muse BV2 mikrogliaceller og musehippocampale HT22-celler med indvirkning på neuroinflammation. Opregulering af HO-1-ekspression via Nrf2-vej i muse BV2 mikrogliaceller, som forsvarer celledød af musehippocampale HT22-celler [38]. Desuden øger kobolt-baserede hybridmolekyler (HYCO'er), der kombinerer en Nrf2-inducer med en frigiver af carbonmonoxid (CO), Nrf2/HO-1-ekspression, frigiver CO og udøver antiinflammatorisk aktivitet in vitro. HYCO'er opregulerer også HO-1 i væv og leverer CO i blodet efter administration in vivo, hvilket understøtter deres potentielle anvendelse mod inflammatoriske tilstande [39]. Nrf2/HO-1 opregulering reducerer inflammation ved at øge den efferocytiske aktivitet af murine makrofager behandlet med taurin chloraminer [40]. Alt i alt afslørede de ovenfor forklarede eksperimentelle modeller, at Nrf2/HO-1-aksen spiller en stor rolle i antiinflammatorisk funktion, hvilket tyder på, at Nrf2 er et terapeutisk mål i inflammationsassocierede sygdomme.
Derudover virker biprodukterne af HO-1, såsom CO, bilirubin, som en kraftig antioxidant under oxidativ stress og celleskader [41], [42]; det undertrykker autoimmun encephalomyelitis og hepatitis [43], [44]; og det beskytter mus og rotter mod endotoksisk shock ved at forhindre dannelsen af iNOS og NO [45], [46], [47]. Desuden reducerer bilirubin endotelaktivering og dysfunktion [48]. Interessant nok reducerer bilirubin transmigrationen af endotelleukocytter via adhæsionsmolekyle-1 [49]. Disse specifikke referencer indikerer, at HO-1 ikke kun virker som et potent anti-inflammatorisk middel, men også dets metabolitter.
Inflammatoriske mediatorer og enzymer hæmmet af Nrf2
Cytokiner og kemokiner
Cytokiner er lavmolekylære proteiner og polypeptider, der udskilles af en række forskellige celler; de regulerer cellevækst, differentiering og immunfunktion og er involveret i inflammation og sårheling. Cytokiner omfatter interleukiner (IL'er), interferoner, tumornekrosefaktor (TNF), kolonistimulerende faktor, kemokiner og vækstfaktorer. Nogle cytokiner regnes som pro-inflammatoriske mediatorer, mens andre har anti-inflammatoriske funktioner. Udsættelse for oxidativ stress resulterer i overproduktion af cytokiner, som forårsager oxidativ stress i målceller. Adskillige pro-inflammatoriske cytokiner overproduceres, når NF-?B aktiveres af oxidativt stress. Ydermere forårsager pro-inflammatorisk oxidativ stress yderligere aktivering af NF-?B og overproduktion af cytokiner. Aktivering af Nrf2/ARE-systemet spiller en vigtig rolle i at forstyrre denne cyklus. Kemokiner er en familie af små cytokiner, hvis hovedrolle er at lede migrationen af inflammatoriske celler. De fungerer hovedsageligt som kemoattraktanter for leukocytter, monocytter, neutrofiler og andre effektorceller.
Det er blevet rapporteret, at aktivering af Nrf2 forhindrer LPS-induceret transkriptionel opregulering af pro-inflammatoriske cytokiner, herunder IL-6 og IL-1? [50]. IL-1? og IL-6-produktion øges også i Nrf2?/? mus med dextransulfat-induceret colitis [51], [52]. Nrf2 hæmmer produktionen af nedstrøms IL-17 og andre inflammatoriske faktorer Th1 og Th17 og undertrykker sygdomsprocessen i en eksperimentel model for multipel sklerose, autoimmun encephalitis [53]. De Nrf2-afhængige antioxidantgener HO-1, NQO-1, Gclc og Gclm blokerer TNF-?, IL-6, monocyt kemo-attraherende protein-1 (MCP1), makrofag-inflammatorisk protein-2 (MIP2) og inflammatorisk mæglere. Men i tilfælde af Nrf2-knockout-mus opstår den antiinflammatoriske effekt ikke [54]. Peritoneale neutrofiler fra Nrf2-knockout-mus behandlet med LPS har signifikant højere niveauer af cytokiner (TNF-a og IL-6) og kemokiner (MCP1 og MIP2) end vildtype (WT) celler [54]. In vitro undertrykker overførsel af Nrf2-genet til humane og kanin-aorta-glatte muskelceller sekretionen af MCP1 [8], [55], og Nrf2-afhængig HO-1-ekspression undertrykker TNF-a-stimuleret NF-?B og MCP-1 sekretion i humane navlevene-endotelceller [56]. Disse resultater antyder, at opregulering af Nrf2-signalering som reaktion på inflammatoriske stimuli hæmmer overproduktionen af pro-inflammatoriske cytokiner og kemokiner samt begrænser aktiveringen af NF-?B.
Celleadhæsionsmolekyler
Celleadhæsionsmolekyler (CAM'er) er proteiner, der binder til celler eller med den ekstracellulære matrix. Placeret på celleoverfladen er de involveret i cellegenkendelse, celleaktivering, signaltransduktion, proliferation og differentiering. Blandt CAM'erne er ICAM-1 og VCAM-1 vigtige medlemmer af immunoglobulinsuperfamilien. ICAM-1 er til stede i lave koncentrationer i leukocyt- og endotelcellemembraner. Ved cytokinstimulering stiger koncentrationen markant. ICAM-1 kan induceres af IL-1 og TNF og udtrykkes af det vaskulære endotel, makrofager og lymfocytter. Det er en ligand for integrin, en receptor fundet på leukocytter. Når ICAM-1-integrinbroen aktiveres, binder leukocytter til endotelceller og migrerer derefter ind i subendotelvæv [57]. VCAM-1 medierer adhæsionen af lymfocytter, monocytter, eosinofiler og basofiler til vaskulært endotel og bidrager til leukocytrekruttering, som i sidste ende fører til vævsskade på grund af oxidativt stress. Nrf2 hæmmer promotoraktiviteten af VCAM-1 [58]. Det Nrf2-regulerede nedstrøms gen HO-1 kan påvirke ekspressionen af E-selectin og VCAM-1, adhæsionsmolekyler forbundet med endotelceller [59]. Lungeekspressionen af flere CAM'er såsom CD-14, TREM1, SELE, SELP og VCAM-1 er signifikant højere i Nrf2?/? mus end i Nrf2+/+ mus [60]. Nrf2 i humane aorta-endotelceller undertrykker TNF-a-induceret VCAM-1-ekspression og interfererer med TNF-a-induceret monocytisk U937-celleadhæsion [8]. Overekspression af Nrf2 hæmmer også TNF-a-induceret VCAM-1-genekspression i humane mikrovaskulære endotelceller [61]. Den naturligt forekommende antioxidant 3-hydroxyanthranilic acid (HA), en af l-tryptophan-metabolitter dannet in vivo langs den metaboliske rute kendt som kynurenin-vejen under inflammation eller infektion, har vist sig at inducere HO-1-ekspression og at stimulere Nrf2 i human navlestreng. vene-endotelceller (HUVEC'er). Nrf2-afhængig HO-1-ekspression induceret af HA hæmmer MCP-1-sekretion, VCAM-1-ekspression og NF-kB-aktivering forbundet med vaskulær skade og inflammation i aterosklerose [56]. Det anti-proliferative og anti-inflammatoriske syntetiske chalconderivat 2a,4a,6a-tris (methoxymethoxy) chalcon hæmmer ICAM-1, det pro-inflammatoriske cytokin IL-1a og TNF-a ekspression i tyktarmsvæv fra mus behandlet med trinitrobenzensulfonsyre [62]. Opregulering af Nrf2 hæmmer den TNF-a-inducerede ICAM-1-ekspression i humane retinale pigmentepitelceller behandlet med lycopen [63]. Alle disse undersøgelser tyder på, at Nrf2 spiller en nøglerolle i den inflammatoriske proces ved at regulere migrationen og infiltrationen af inflammatoriske celler til betændt væv.
Matrix metalloproteinaser (MMP'er)
MMP'er er bredt til stede i den ekstracellulære matrix og er involveret i fysiologiske og patologiske processer såsom celleproliferation, migration, differentiering, sårheling, angiogenese, apoptose og tumormetastase. Det er blevet rapporteret, at Nrf2/HO-1-aksen hæmmer MMP-9 i makrofager og MMP-7 i humane tarmepitelceller, og dette er gavnligt i behandlingen af inflammatorisk tarmsygdom [62], [64]. UV-bestråling-induceret hudskade er mere alvorlig i Nrf2-knockout end i WT-mus, og MMP-9-niveauet er signifikant højere, hvilket indikerer, at Nrf2 reducerer MMP-9-ekspression. Derfor anses Nrf2 for at være beskyttende mod UV-bestråling [65]. En anden undersøgelse rapporterede også, at den nedregulerede transkriptionelle aktivering af MMP-9 i tumorcelleinvasion og -inflammation reguleres gennem hæmning af NF-kB-signalvejen [66]. Ved traumatisk rygmarvsskade deltager NF-kB-signalvejen også i reguleringen af mRNA-niveauerne af MMP-9 [67]. Derfor påvirkes reguleringen af MMP'er ved inflammation direkte af Nrf2-vejen eller indirekte gennem den Nrf2-påvirkede NF-?B-vej.
Cyclooxygenase-2 (COX2) og inducerbar nitrogenoxidsyntase (INOS)
En række eksperimenter på Nrf2-knockout-mus har vist dens afgørende rolle i inflammation og reguleringen af pro-inflammatoriske gener såsom COX-2 og iNOS. For første gang har Khor et al. rapporterede øget ekspression af pro-inflammatoriske cytokiner såsom COX-2 og iNOS i tyktarmsvævene i Nrf2?/? mus sammenlignet med WT Nrf2+/+ mus, hvilket indikerer, at Nrf2 undertrykker deres aktivitet [51]. En anden rapport om forbehandling med sulforaphane, en af de velkendte Nrf2-aktivatorer til stede i korsblomstrede grøntsager, demonstrerede dens anti-inflammatoriske virkning ved at hæmme ekspressionen af TNF-?, IL-1?, COX-2 og iNOS ved både mRNA'et og proteinniveauer i primære peritoneale makrofager fra Nrf2+/+ mus sammenlignet med dem fra Nrf2?/? mus [68]. Tilsvarende viser hippocampus af Nrf2-knockout-mus med LPS-induceret inflammation også højere ekspression af inflammationsmarkører såsom iNOS, IL-6 og TNF-? end WT-mus [69]. Ligeledes er Nrf2-knockout-mus overfølsomme over for det oxidative stress induceret af 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin samt viser øgede mRNA- og proteinniveauer af inflammationsmarkører såsom COX-2, iNOS IL-6 og TNF-a [70]. Desuden lever fra Nrf2?/? mus udfordret med en methionin- og cholin-mangel kost har ~ 5 gange højere mRNA-ekspression af Cox2 og iNOS end dem fra WT-mus på samme diæt, hvilket tyder på en anti-inflammatorisk rolle af Nrf2 [71]. For nylig har Kim et al. demonstreret, at det fytokemiske ethylpyruvat udøver sine antiinflammatoriske og antioxidative virkninger ved at reducere ekspressionen af iNOS gennem Nrf2-signalering i BV2-celler. De viste, at ethylpyruvat inducerer nuklear translokation af Nrf2, som i sidste ende hæmmer interaktionen mellem p65 og p300, hvilket fører til nedsat ekspression af iNOS [72]. Ydermere aktiverer carbazolanalogen LCY-2-CHO Nrf2 og forårsager dens nukleare translokation, hvilket fører til undertrykkelse af COX2 og iNOS-ekspression [73] i rotte aorta vaskulære glatte muskelceller.
Paradoksal rolle for Nrf2 i reguleringen af NLRP3 iIflammasome�aktivitet
NLR-familien, pyrindomæne indeholdende 3 (NLRP3) inflammasom er et multiproteinkompleks, der fungerer som en patogengenkendelsesreceptor (PRR) og genkender det brede udvalg af mikrobielle, oxidative stresssignaler såsom patogenassocierede molekylære mønstre (PAMP'er), skade- associerede molekylære mønstermolekyler (DAMP'er) og ROS [74]. Det aktiverede NLRP3-inflammasom medierer spaltningen af caspase-1 og sekretion af pro-inflammatorisk cytokin interleukin-1? (IL-1?), der i sidste ende inducerer celledødsprocessen kendt som pyroptose, der beskytter værter mod en lang række patogener [75]. Imidlertid er afvigende aktivering af inflammasomet forbundet med proteinfejlfoldningssygdomme såsom overførbare spongiforme encephalopatier, Alzheimers sygdom, Parkinsons sygdom og også type 2 diabetes [76], cancer [77], gigt og åreforkalkning [78].
En nylig observation fra Rong Hu-gruppen om association af Nrf2 med negativ regulering af inflammasom afslørede, at Nrf2 inducerer NQO1-ekspressionen, der fører til inhibering af NLRP3-inflammasomaktivering, caspase-1-spaltning og IL-1? generering i makrofager. Desuden regulerede en velkendt Nrf2-aktivator, tert-butylhydroquinon (tBHQ), negativt reguleret NLRP3-transkription ved at aktivere ARE på Nrf2-afhængig måde [79]. Ud over ovenstående observation er den samme gruppe også blevet afsløret, at dimethylfumarat (DMF) forhindrer DSS-induceret colitis via aktiverende Nrf2-signalvej, som er involveret i Nrf2-nuklear translokation og inhibering af NLRP3-inflammasomsamling [80].
En række eksperimenter med naturlige og syntetiske forbindelser har også afsløret den hæmmende effekt af Nrf2 på NLRP3-inflammasomaktivering. For eksempel har behandling af epigallocatechin-3-gallat (EGCG) i lupus nefritis-mus vist sig at reducere nyre NLRP3-inflammasomaktivering, som medieres af Nrf2-signalvej [81]. Ligeledes hæmmer citral (3,7-dimethyl-2,6-octadienal), en vigtig aktiv forbindelse i en kinesisk urtemedicin Litsea cubeba, NLRP3-inflammasomaktiveringen via Nrf2-antioxidantsignalvejen i musemodellen Accelerated and Severe Lupus Nephritis (ASLN) [82]. På samme måde beskyttede biochanin mod LPS/GalN-induceret leverskade ved at aktivere Nrf2-vejen og hæmme NLRP3-inflammasomaktivering i BALB/c-hanmus [83]. Endvidere blev mangiferin også vist at opregulere ekspressionen af Nrf2 og HO-1 på en dosisafhængig måde og hæmmede LPS/D-GalN-induceret hepatisk NLRP3, ASC, caspase-1, IL-1? og TNF-? udtryk [84].
På trods af den negative regulering af NLRP3 af Nrf2, aktiverer den også NLRP3 og AIM2 inflammasomfunktionen. Haitao Wen og kolleger opdagede, at Nrf2 ?/? musemakrofager har vist den defekte aktivering af NLRP3- og AIM2-inflammasomet, men ikke NLRC4-inflammasomet [85]. Interessant nok skildrer denne observation de ukendte funktioner af Nrf2 i sammenhæng med betændelsesassocierede sygdomme; derfor er det meget vigtigt at studere yderligere for at afsløre mekanismen, hvori Nrf2 aktiverer inflammasomfunktionen, før man betragter det som et terapeutisk mål.
Undertrykkelse af pro-inflammatorisk cytokintransskription af Nrf2
En meget nylig undersøgelse baseret på kromatin-immunpræcipitation (ChIP)-seq og ChIP-qPCR-resultater i musemakrofager afslørede, at Nrf2 binder til pro-inflammatoriske cytokiner som IL-6 og IL-1? og hæmmer RNA Pol II-rekruttering. Som et resultat er RNA Pol II ikke i stand til at behandle den transkriptionelle aktivering af IL-6 og IL-1? som i sidste ende fører til hæmning af genekspression. For første gang afslørede Masayuki Yamamotos gruppe den nye mekanisme, hvorved Nrf2 ikke kun transaktiverer sine nedstrømsgener gennem ARE'er, men også undertrykker den transkriptionelle aktivering af specifikke gener med eller uden en ARE ved at hæmme rekrutteringen af RNA Pol II [50].
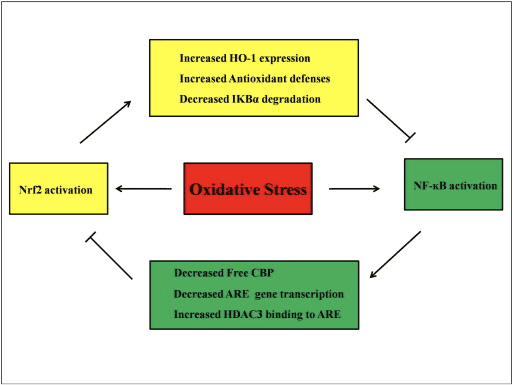
Krydstale mellem Nrf2 og NF-?B Pathways
NF-?B er et proteinkompleks, der er ansvarligt for DNA-transkription, der findes i næsten alle typer dyreceller og er involveret i forskellige processer såsom inflammation, apoptose, immunrespons, cellevækst og udvikling. p65, et Rel-protein fra NF-aB-familien, har et transaktiveringsdomæne, hvorimod p50 ikke og kræver heterodimerisering med Rel-protein for at aktivere transkription. Under oxidativ stress aktiveres I?B-kinase (IKK) og forårsager phosphorylering af I?B, hvilket resulterer i frigivelse og nuklear translokation af NF-?B. NF-aB forårsager transkription af pro-inflammatoriske mediatorer såsom IL-6, TNF-a, iNOS, IL-1 og intracellulær adhæsion COX-2.
Unormal regulering af NF-?B er blevet forbundet med rheumatoid arthritis, astma, inflammatorisk tarmsygdom og Helicobacter pylori-infektion-induceret gastritis [86]. Det vurderes i øjeblikket, at NF-kB-aktivitet påvirker Keapl/Nrf2/ARE-signalvejen hovedsageligt i tre aspekter: For det første nedbryder Keap1 IKK? gennem ubiquitinering, hvilket hæmmer aktiviteten af NF-?B [87]. For det andet inducerer den inflammatoriske proces inflammatoriske mediatorer som COX2 afledt af cyclopentenon prostaglandin 15d-PGJ2, en stærk elektrofil, der reagerer med Keap1 og aktiverer Nrf2, og dermed initierer gentranskription med samtidig hæmning af NF-kB aktivitet [58], [88] ( Fig. 3 A, B). For det tredje kan NF-?B kombineres med den konkurrerende Nrf2 transkriptionelle co-aktivator CBP [89], [90] (Fig. 3 C, D).
Figur 3 Krydstale mellem Nrf2- og NF-?B-vejene. (A) Keap1 dirigerer IKK til CUL3-medieret ubiquitinering og proteasomnedbrydning, som i sidste ende fører til inhibering af NF-?B-phosphorylering, og denne mekanisme fungerer også som kompetitiv binding af Nrf2 og IKK med Keap1. (B) Oxidativt stress aktiverer IKK, som phosphorylerer NF-?B, hvilket fører til dets translokation ind i kernen og aktivering af proinflammatoriske cytokiner såsom COX-2. Det terminale produkt af COX-2 kendt som 15d-PGJ2 fungerer som en inducer af Nrf2, der i sidste ende fører til undertrykkelse af oxidativt stress. (C) Nrf2 binder med sin transkriptionelle cofaktor CBP sammen med små Maf og andre transkriptionelle maskineri for at initiere ARE-drevet genekspression. (D) Når NF-?B binder til CBP på en kompetitiv måde, hæmmer det bindingen af CBP med Nrf2, hvilket fører til inhibering af Nrf2-transaktivering.
Det antages, at Nrf2- og NF-?B-signalvejene interagerer for at kontrollere transkriptionen eller funktionen af nedstrøms målproteiner. Som begrundelse for denne antagelse viser mange eksempler, at direkte eller indirekte aktivering og inhibering forekommer mellem medlemmer af Nrf2- og NF-?B-vejene (fig. 4). Som svar på LPS øger Nrf2-knockdown signifikant NF-?B-transkriptionsaktiviteten og NF-?B-afhængig gentranskription, hvilket viser, at Nrf2 hæmmer NF-?B-aktivitet [60], [91]. Derudover hæmmer øget ekspression af Nrf2-afhængig nedstrøms HO-1 NF-?B-aktivitet. Når prostatacancerceller kortvarigt udsættes for β-tochopherylsuccinat, et derivat af vitamin E, opreguleres HO-1-ekspression. Slutprodukterne af HO-1 hæmmer den nukleare translokation af NF-?B [92]. Disse in vivo undersøgelser tyder på, at Nrf2 negativt regulerer NF-kB signalvejen. LPS stimulerer NF-?B DNA-bindingsaktivitet, og niveauet af p65-underenheden af NF-?B er signifikant højere i nukleare ekstrakter fra lungerne af Nrf2?/? end fra WT-mus, hvilket tyder på en negativ rolle af Nrf2 i NF-?B-aktivering. Desuden Nrf2?/? museembryofibroblaster behandlet med LPS og TNF-? viser mere fremtrædende NF-?B-aktivering forårsaget af IKK-aktivering og I?B-? nedbrydning [60]. Og clearance af respiratorisk syncytialvirus er signifikant reduceret, mens NF-?B DNA-bindingsaktivitet er øget i Nrf2?/? mus sammenlignet med WT-mus [93]. Pristan-induceret lupus nefritis i Nrf2?/? mus behandlet med sulforaphane har alvorlig nyreskade og patologiske ændringer såvel som forhøjet iNOS-ekspression og NF-?B-aktivering sammenlignet med WT, hvilket tyder på, at Nrf2 forbedrer lupus nefritis ved at hæmme NF-?B-signalvejen og rydde ROS [94] ]. NF-?B-aktivitet forekommer også, når celler behandles med en Nrf2-inducer sammen med LPS og TNF-?. For eksempel inhiberer et syntetisk chalcone-derivat TNF-a-induceret NF-aB-aktivering både direkte og indirekte og delvist gennem induktionen af HO-1-ekspression i humane intestinale HT-29-celler [62]. Undertrykkelse af NF-?B-translokation og DNA-bindingsaktivitet samt undertrykkelse af iNOS-ekspression i hepatocytter findes, når F344-rotter behandles med 3H-1,2-dithiol-3-thion (D3T) [95]. Efter samtidig behandling med sulforaphane og LPS, den LPS-inducerede ekspression af iNOS, COX-2 og TNF-? i Raw 264.7 er makrofager nedreguleret, hvilket tyder på, at sulforaphan har anti-inflammatorisk aktivitet via inhibering af NF-?B DNA-binding [96]. Selvom flere eksperimentelle undersøgelser er blevet udført for at forklare sammenhængen mellem Nrf2- og NF-?B-vejene, er der stadig modstridende resultater. Både positive og negative reguleringer er blevet rapporteret mellem Nrf2 og NF-kB [97]. Sædvanligvis aktiverer kemoforebyggende elektrofiler 3H-1,2-dithiole-3-thion, sulforaphane og Triterpenoid CDDO-Me Nrf2 ved at hæmme NF-kB og dets nedregulerede gener [98], [99], [100]. I modsætning hertil har flere midler eller tilstande såsom ROS, LPS, flow shear stress, oxideret LDL og cigaretrøg vist sig at øge både Nrf2 og NF-kB aktivitet [97]. Derudover har in vivo undersøgelser afsløret, at NF-kB aktivitet er nedsat i lever isoleret fra Nrf2?/? mus og NF-?B-bindingsaktivitet er lavere i Nrf2a/? end i Nrf2+/+ mus [101]. Imidlertid hæmmer humane aorta-endotelceller behandlet med adenoviral vektor Nrf2 NF-?B nedstrøms gener uden at påvirke aktiviteten af NF-?B [8].
Figur 4 Regulatorisk sløjfe af Nrf2 og NF-?B. Nrf2-vejen hæmmer NF-?B-aktivering ved at forhindre nedbrydning af I?B-? og øget HO-1-ekspression og antioxidantforsvar, som neutraliserer ROS og afgiftende kemikalier. Som et resultat undertrykkes ROS-associeret NF-?B-aktivering. Ligeledes reducerer NF-?B-medieret transkription Nrf2-aktivering ved at reducere�ER�gentransskription og frit CREB-bindende protein ved at konkurrere med Nrf2 om CBP. Desuden øger NF-?B rekrutteringen af histon-deacetylase (HDAC3) til ARE-regionen, og derfor forhindres Nrf2-transkriptionel aktivering.
Aktiveringen af Nrf2-signalvejen spiller en stor rolle i ekspressionen af enzymer og gener involveret i afgiftningen af reaktive oxidanter ved at forbedre antioxidantkapaciteten i cellerne i den menneskelige krop. Mens mange forskningsundersøgelser er tilgængelige i dag, er de regulatoriske mekanismer i Nrf2-aktivering ikke fuldt ud forstået. En mulig rolle for Nrf2-signalvejen i behandlingen af inflammation er også blevet fundet. Dr. Alex Jimenez DC, CCST Insight
Rolle af Nrf2 i inflammatoriske sygdomme
In vivo undersøgelser har vist, at Nrf2 spiller en vigtig rolle i inflammatoriske sygdomme, der påvirker forskellige systemer; disse omfatter gastritis, colitis, gigt, lungebetændelse, leverskader, hjerte-kar-sygdomme, neurodegenerative sygdomme og hjerneskade. I disse undersøgelser har Nrf2?/? dyr viste mere alvorlige symptomer på betændelse og vævsskade end WT-dyr. Derfor menes det, at Nrf2-signalvejen har en beskyttende effekt ved inflammatoriske sygdomme. Intratracheal installation af porcin pancreaselastase inducerer kronisk obstruktiv lungesygdom, især emfysem. Nrf2-deficiente mus er meget modtagelige for emfysem, og nedsat ekspression af HO-1, PrxI og antiproteasegenet SLPI forekommer i alveolære makrofager. Nrf2 anses for at være en nøgleregulator i det makrofagmedierede forsvarssystem mod lungeskade [102]. Nrf2-mangelfulde mus med emfysem induceret af tobaksrøgeksponering i 6 måneder viser øget bronkoalveolær inflammation, opreguleret ekspression af oxidative stressmarkører i alveoler og øget alveolær septalcelle-apoptose, hvilket tyder på, at Nrf2 virker mod tobaksinduceret emfysem gennem den øgede ekspression af antioxidant gener [102], [103]. Med Nrf2-afbrydelse viser allergenmedieret luftvejsbetændelse og astma ved brug af ovalbuminkompleks øget luftvejsinflammation, luftvejshyperreaktivitet, hyperplasi af bægerceller og høje niveauer af Th2 i bronkoalveolær lavage og splenocytter, hvorimod den Nrf2-medierede signalvej begrænser luftvejs eosinofili , slim hypersekretion og luftvejshyperreaktivitet samt inducering af mange antioxidantgener, der forhindrer udviklingen af astma [104]. Carrageenaninjektion i pleurahulen inducerer pleuritis, og 15d-PGJ2-akkumulering i Nrf2-inflammatoriske celler er begrænset til muse-peritoneale makrofager. I den tidlige fase af inflammation aktiverer 15d-PGJ2 Nrf2 og regulerer den inflammatoriske proces via induktion af HO-1 og PrxI. En undersøgelse antydede også, at COX-2 har en anti-inflammatorisk effekt i den tidlige fase ved produktion af 15d-PGJ2 [105]. Oral administration af 1% dextransulfat-natrium i 1 uge inducerer colitis forbundet med histologiske ændringer, der omfatter forkortelse af krypter og infiltration af inflammatoriske celler i tyktarmsvæv. For at beskytte tarmens integritet i colitis kunne Nrf2 spille en vigtig rolle ved at regulere pro-inflammatoriske cytokiner og inducere fase II afgiftende enzymer [51]. I en Nrf2-knockout musemodel af LPS-induceret pulmonal sepsis regulerer NF-?B aktivitet indflydelsen af inflammatoriske cytokiner såsom COX-2, IL-113, IL-6 og TNF? som er afgørende for at starte og fremme inflammation [60]. Nrf2 reducerer inflammatorisk skade ved at regulere disse inflammatoriske faktorer. I disse modeller af akut inflammation reducerer den øgede regulering af antioxidantenzymer, pro-inflammatoriske cytokiner og mediatorer ved Nrf2-signalvejen den inflammatoriske skade i WT-dyr. Interessant nok er dette også blevet rapporteret i Nrf2-knockout-mus, hvor symptomerne er markant forværret sammenlignet med WT-mus.
Forskning i Nrf2-afhængige antiinflammatoriske lægemidler
Sammenfattende har vi diskuteret eksperimenter, der viser, at Nrf2-signalvejen spiller en regulerende rolle i mange områder af inflammation, så Nrf2-afhængige antiinflammatoriske midler er vigtige til behandling af inflammatoriske sygdomme.
Planter har været ekstraordinært rige kilder til forbindelser, der aktiverer Nrf2-transkriptionsfaktor, hvilket fører til opregulering af cytobeskyttende gener. For nylig blev der udført adskillige undersøgelser for at undersøge virkningerne af forskellige antiinflammatoriske midler, for det meste af vegetabilsk oprindelse. For eksempel er curcumin den aktive ingrediens i gurkemeje og findes også i små mængder i ingefær; isothiocyanater, specifikt phenylisothiocyanater, er fra broccoli, selleri og andre grøntsager; og anthocyaniner er fra bær og druer [124]. Undersøgelser har vist, at alle disse midler ikke kun er gode antioxidanter, men også har potente antiinflammatoriske virkninger via Nrf2-induktion [125], [126]. Derfor har udviklingen af nye antiinflammatoriske Nrf2-aktivatorer fra planteekstrakt tiltrukket sig stor interesse i medicinsk forskning.
I de senere år er mange dyreforsøg blevet udført for at bekræfte virkningerne af disse forbindelser. Artesunate bruges hovedsageligt til svær malaria, cerebral malaria og reumatiske autoimmune sygdomme; det er også effektivt ved septisk lungeskade. Artesunate aktiverer Nrf2- og HO-1-ekspression, og sidstnævnte reducerer tilstrømningen af pro-inflammatoriske cytokiner og leukocytter til væv for at forhindre inflammation [127]. Isovitexin, udvundet fra skrogerne af Oryza sativa ris, menes at have anti-inflammatoriske og antioxidante egenskaber; det spiller en beskyttende rolle mod LPS-induceret akut lungeskade ved at aktivere Nrf2/HO-1-vejen og hæmme MAPK og NF-?B [128]. Fimasartan, en nyligt populær angiotensin II-receptorblokker, der virker på renin-angiotensin-systemet, reducerer blodtrykket; Brug af fimasartan til at behandle mus med kirurgisk induceret unilateral ureteral obstruktion reducerer oxidativt stress, inflammation og fibrose via opregulering af Nrf2 og antioxidantvejen og hæmning af RAS og MAPK'er [129]. Sappanone er vidt udbredt i Sydøstasien, hvor det bruges som en anti-influenza, anti-allergisk og neurobeskyttende medicin; det aktiverer Nrf2 og hæmmer NF-?B og kan derfor være gavnligt i behandlingen af Nrf2- og/eller NF-?B-relaterede sygdomme [130]. Bixin udvundet af frøene af Bixin orellana bruges til infektions- og inflammatoriske sygdomme i Mexico og Sydamerika; det reducerer inflammatoriske mediatorer, alveolær kapillær lækage og oxidativ skade på en Nrf2-afhængig måde for at lindre ventilationsinduceret lungeskade og genoprette normal lungemorfologi [131]. Andre planteforbindelser, såsom epigallocatechin gallat, sulforaphane, resveratrol, lycopen og grøn te-ekstrakt har terapeutiske virkninger på inflammatoriske sygdomme gennem Nrf2-signalvejen [132], [133], [134]. For nylig er et andet fytokemikalie, eriodictyol, som er til stede i citrusfrugter, blevet rapporteret at have anti-inflammatoriske og antioxidante virkninger på cisplatin-induceret nyreskade og sepsis-induceret akut lungeskade ved at regulere Nrf2, hæmme NF-?B og hæmme ekspression af cytokiner i makrofager [135], [136]. Imidlertid viser talrige fytokemikalier et stort lovende for forebyggelse og behandling af forskellige menneskelige sygdomme, og nogle er allerede gået ind i kliniske forsøgsstadiet (tabel 2).
Disse planteforbindelser aktiverer Nrf2-signalvejen hovedsageligt i form af elektrofile materialer, der modificerer cysteinresterne af Keap1, hvilket fører til fri nuklear Nrf2-binding med ARE, hvilket resulterer i aktivering af transkription af det tilsvarende gen.
Sulforaphane og dens virkninger på kræft, dødelighed, aldring, hjerne og adfærd, hjertesygdomme og mere
Isothiocyanater er nogle af de vigtigste planteforbindelser, du kan få i din kost. I denne video gør jeg det mest omfattende tilfælde til dem, der nogensinde er blevet lavet. Kort opmærksomhed span? Spring til dit yndlingsemne ved at klikke på et af nedenstående tidspunkter. Fuld tidslinje nedenfor.
Nøglesektioner:
00: 01: 14 - Kræft og dødelighed
00: 19: 04 - Aging
00: 26: 30 - Hjerne og adfærd
00: 38: 06 - Final recap
00: 40: 27 - Dosis
Fuld tidslinje:
00: 00: 34 - Introduktion af sulforaphane, et vigtigt fokus i videoen.
00: 01: 14 - Cruciferous vegetabilsk forbrug og reduktion af allårsag dødelighed.
00: 02: 12 - Prostatacancerrisiko.
00: 02: 23 - Kræftrisiko.
00: 02: 34 - Lungekræft i rygere risiko.
00: 02: 48 - Brystkræftrisiko.
00: 03: 13 - Hypotetisk: Hvad hvis du allerede har kræft? (Interventionel)
00: 03: 35 - Plausibel mekanisme, der driver kræft- og dødelighedsassosierende data.
00: 04: 38 - Sulforaphane og kræft.
00: 05: 32 - Dyrebevis, der viser stærk virkning af broccoli-spireekstrakt på udviklingen af blæretumor hos rotter.
00: 06: 06 - Virkning af direkte tilsætning af sulforaphan hos patienter med prostatacancer.
00: 07: 09 - Bioakkumulering af isothiocyanatmetabolitter i egentligt brystvæv.
00: 08: 32 - Inhibering af brystcancerstamceller.
00: 08: 53 - Historie lektion: brassicas blev etableret som at have sundhedsegenskaber selv i det gamle Rom.
00: 09: 16 - Sulforaphans evne til at øge kræftfremkaldende udskillelse (benzen, acrolein).
00: 09: 51 - NRF2 som en genetisk switch via antioxidant responselementer.
00: 10: 10 - Hvordan NRF2 aktivering øger kræftfremkaldende udskillelse via glutathion-S-konjugater.
00: 10: 34 - Spiraler øger glutathion-S-transferase og reducerer DNA-beskadigelse.
00: 11: 20 - Broccoli-spire drikker øger benzenudskillelsen med 61%.
00: 13: 31 - Broccoli spirehomogenat øger antioxidant enzymer i den øvre luftvej.
00: 15: 45 - Cruciferous vegetabilsk forbrug og dødelighed i hjertesygdomme.
00: 16: 55 - Broccoli spire pulver forbedrer blodlipider og overordnet hjertesygdomsrisiko hos type 2 diabetikere.
00: 19: 04 - Begyndelse af aldringssektionen.
00: 19: 21 - Sulforaphane beriget kost forbedrer levetiden for biller fra 15 til 30% (under visse forhold).
00: 20: 34 - Betydningen af lav inflammation for lang levetid.
00: 22: 05 - Cruciferous grøntsager og broccoli spire pulver synes at reducere et bredt udvalg af inflammatoriske markører hos mennesker.
00: 24: 14 - Musestudier tyder på, at sulforaphan kan forbedre adaptiv immunfunktion i alderdommen.
00: 25: 18 - Sulforaphane forbedrede hårvækst i en musmodel af skaldethed. Billede på 00: 26: 10.
00: 26: 30 - Begyndelse af hjerne- og adfærdssektionen.
00: 27: 18 - Effekt af broccoli-spireekstrakt på autisme.
00: 27: 48 - Virkning af glucoraphanin på skizofreni.
00: 28: 17 - Start af depression diskussion (sandsynlig mekanisme og undersøgelser).
00: 31: 21 - Mus undersøgelse ved hjælp af 10 forskellige modeller af stress-induceret depression viser sulforaphan på samme måde som fluoxetin (prozac).
00: 32: 00 - Undersøgelse viser direkte indtagelse af glucoraphanin hos mus er tilsvarende effektiv til forebyggelse af depression fra social nederlagsspændingsmodel.
00: 33: 01 - Begyndelsen af neurodegenerationsafsnittet.
00: 33: 30 - Sulforaphane og Alzheimers sygdom.
00: 33: 44 - Sulforaphane og Parkinsons sygdom.
00: 33: 51 - Sulforaphane og Hungtington's sygdom.
00: 35: 55 - Sulforaphane og neuronal plasticitet.
00: 36: 32 - Sulforaphane forbedrer læring som model af type II diabetes hos mus.
00: 37: 19 - Sulforaphane og duchenne muskeldystrofi.
00: 37: 44 - Myostatininhibering i muskel-satellitceller (in vitro).
00: 38: 06 - Late video recap: dødelighed og kræft, DNA-beskadigelse, oxidativ stress og betændelse, udslip af benzen, kardiovaskulær sygdom, type II diabetes, hjernens virkninger (depression, autisme, skizofreni, neurodegeneration), NRF2-pathway.
00: 40: 27 - Tanker om at finde ud af en dosis broccolispirer eller sulforaphane.
00: 41: 01 - Anecdoter på spiring hjemme.
00: 43: 14 - På tilberedningstemperaturer og sulforafanaktivitet.
00: 43: 45 - Gut-bakteriekonvertering af sulforaphan fra glucoraphanin.
00: 44: 24 - Tilskud fungerer bedre, når de kombineres med aktiv myrosinase fra grøntsager.
00: 44: 56 - Madlavningsteknikker og cruciferous grøntsager.
00: 46: 06 - Isothiocyanater som goitrogener.
konklusioner
I øjeblikket har meget forskning fokuseret på rollen som Nrf2/Keap1/ARE-signalvejen i inflammation. Blandt de enzymer, der opreguleres af Nrf2, er HO-1 et af de repræsentative stressresponsenzymer. HO-1 har fremtrædende antiinflammatoriske og antioxidante egenskaber. Generelt regulerer Nrf2-signalvejen også negativt cytokiner, kemokinfrigivende faktorer, MMP'er og andre inflammatoriske mediatorer COX-2- og iNOS-produktion, som direkte eller indirekte påvirker de relevante NF-kB- og MAPK-veje og andre netværk, der kontrollerer inflammation. Det foreslås, at Nrf2- og NF-?B-signalvejene interagerer for at regulere transkriptionen eller funktionen af nedstrøms målproteiner. Undertrykkelse eller inaktivering af NF-?B-medieret transkriptionel aktivitet gennem Nrf2 forekommer højst sandsynligt i den tidlige fase af inflammation, da NF-?B regulerer de novo-syntesen af en række pro-inflammatoriske mediatorer. Der er dog stadig nogle begrænsninger i forskningen, såsom om der er sammenhænge mellem Nrf2 og andre signalveje såsom JAK/STAT, betydningen af de nuværende Nrf2-aktivatorer afledt af naturlige plantekilder i inflammation, og hvordan man kan forbedre den biologiske aktivitet og forbedre målretningen af disse forbindelser. Disse kræver yderligere eksperimentel validering.
Derudover kan Nrf2-signalvejen regulere > 600 gener [163], hvoraf > 200 koder for cytobeskyttende proteiner [164], der også er forbundet med inflammation, cancer, neurodegenerative sygdomme og andre større sygdomme [165]. Voksende beviser, der tyder på, at Nrf2-signalvejen er dereguleret i mange kræftformer, hvilket resulterer i afvigende ekspression af Nrf2-afhængigt genbatteri. Desuden spiller betændelse en stor rolle i oxidativt stress-relaterede sygdomme, især i cancer. Anvendelse af flere Nrf2-aktivatorer for at modvirke inflammationen kan resultere i afvigende ekspression af Nrf2-gener, som inducerer onkogenese og resistens over for kemo- og/eller radioterapi. Derfor kan meget specifikke aktivatorer af Nrf2 udvikles for at minimere dets pleiotrope virkninger. Adskillige aktivatorer af Nrf2 har vist en signifikant forbedring af de antiinflammatoriske funktioner i oxidativt stress-relaterede sygdomme. Det bedste eksempel på Nrf2-aktivator godkendt af FDA og almindeligt anvendt til behandling af inflammatoriske sygdomme som multipel sklerose (MS) er dimethylfumarat. Tecfidera� (registreret navn på dimethylfumarat af Biogen) anvendes effektivt til behandling af recidiverende former for multipel sklerose hos et stort antal patienter [152]. Effektiviteten af at bruge Nrf2-aktivatorer til behandling af inflammatoriske sygdomme kræver imidlertid yderligere validering for at undgå de skadelige virkninger af Nrf2. Derfor kan udvikling af terapier til anti-inflammationsaktiviteten medieret af Nrf2 have betydelig klinisk effekt. Igangværende undersøgelser af Nrf2-signalvejen rundt om i verden er afsat til at udvikle meget målrettede terapeutiske midler til at kontrollere symptomerne på inflammation og til at forebygge og behandle kræft såvel som neurodegenerative og andre større sygdomme.
Afslutningsvis fornemmer Nrf2 niveauerne af oxidativt stress i den menneskelige krop og hjælper i sidste ende med at fremme reguleringen af antioxidanter og afgiftende enzymer og gener. Fordi kronisk inflammation forårsaget af øgede niveauer af oxidativt stress er blevet forbundet med neurodegenerative sygdomme, Nrf2 kan spille en væsentlig rolle i behandling af sundhedsproblemer som Alzheimers sygdom, blandt andre. Omfanget af vores information er begrænset til kiropraktik og rygsøjlesundhedsproblemer. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Yderligere emne diskussion: Relieving knæsmerter uden kirurgi
Knæsmerter er et velkendt symptom, der kan opstå på grund af en række knæskader og / eller tilstande, herunder sportsskader. Knæet er et af de mest komplekse led i den menneskelige krop, da det består af skæringspunktet mellem fire knogler, fire ledbånd, forskellige sener, to menisker og brusk. Ifølge American Academy of Family Physicians omfatter de mest almindelige årsager til knæsmerter patellar subluksation, patellar tendinitis eller jumper's knee og Osgood-Schlatters sygdom. Selvom knæsmerter mest sandsynligt forekommer hos personer over 60 år, kan knæsmerter også forekomme hos børn og unge. Knæsmerter kan behandles hjemme efter RICE-metoderne, dog kan alvorlige knæskader kræve øjeblikkelig lægehjælp, herunder kiropraktisk behandling. �
Neurodegenerative sygdomme, såsom Alzheimers sygdom og Parkinsons sygdom, påvirker millioner af individer verden over. En række forskellige behandlingsmuligheder er tilgængelige for at behandle symptomerne på flere neurodegenerative sygdomme, selvom resultaterne ofte er begrænsede. Forskningsundersøgelser har fundet ud af, at oxidativ stress forårsaget af både interne og eksterne faktorer kan være en årsag til udviklingen af neurodegenerative sygdomme. Det transskriptionsfaktor, Nrf2, er blevet bestemt til at fungere som en vigtig forsvarsmekanisme mod oxidativt stress. Formålet med artiklen nedenfor er at vise effekterne af Nrf2 om neurodegenerative sygdomme.
Modulation af proteostase ved transkriptionsfaktor NRF2
Neurodegenerative sygdomme er forbundet med akkumulering af specifikke proteinaggregater, hvilket tyder på en intim forbindelse mellem skadet hjerne og tab af proteostase. Proteostase refererer til alle de processer, hvorved celler kontrollerer overfloden og foldningen af proteomet takket være et bredt netværk, der integrerer reguleringen af signalveje, genekspression og proteinnedbrydningssystemer. Denne gennemgang forsøger at opsummere de mest relevante resultater om den transkriptionelle modulering af proteostase, der udøves af transkriptionsfaktoren NRF2 (nuklear faktor (erythroid-afledt 2)-lignende 2). NRF2 er klassisk blevet betragtet som hovedregulatoren af antioxidantcelleresponset, selvom det i øjeblikket fremstår som en nøglekomponent i transduktionsmaskineriet til at opretholde proteostase. Som vi vil diskutere, kunne NRF2 forestilles som et knudepunkt, der kompilerer nødsignaler afledt af fejlfoldet proteinakkumulering for at opbygge en koordineret og holdbar transkriptionel respons. Dette opnås ved funktioner af NRF2 relateret til styring af gener involveret i vedligeholdelsen af den endoplasmatiske retikulums fysiologi, proteasomet og autofagi.
Nuclear Factor (erythroid-afledt 2)-lignende 2 (NRF2) er et basis-leucin-lynlås-protein, der i dag betragtes som en masterregulator af cellulær homeostase. Det kontrollerer den basale og stress-inducerbare ekspression af over 250 gener, der har fælles en cis-virkende forstærker kaldet antioxidant respons element (ARE) [1], [2], [3], [4], [5]. Disse gener deltager i fase I, II og III afgiftningsreaktioner, glutathion og peroxiredoxin/thioredoxin metabolisme, NADPH-produktion gennem pentosephosphat-vejen og æblesyreenzymet, fedtsyreoxidation, jernmetabolisme og proteostase [6]. I betragtning af disse brede cytobeskyttende funktioner er det muligt, at et enkelt farmakologisk hit i NRF2 kan afbøde virkningen af de vigtigste syndere i kroniske sygdomme, herunder oxidativ, inflammatorisk og proteotoksisk stress. NRF2's rolle i moduleringen af antioxidantforsvaret og opløsning af inflammation er blevet behandlet i adskillige undersøgelser (gennemgået i [7]). Her vil vi fokusere på dens rolle i proteostase, dvs. den homøostatiske kontrol af proteinsyntese, foldning, trafficking og nedbrydning. Eksempler vil blive givet i forbindelse med neurodegenerative sygdomme.
Tab af proteostase påvirker NRF2-aktivitet i neurodegenerative sygdomme
Et generelt kendetegn ved neurodegenerative sygdomme er forekomsten af afvigende aggregering af nogle proteiner. Fejlfoldede proteinaggregater af β-synuklein (β-SYN) findes således i Parkinsons sygdom (PD), β-amyloid (Aβ) plaques og hyperfosforylerede TAU neurofibrillære sammenfiltringer ved Alzheimers sygdom (AD), huntingtin (Htt) i Huntingtons sygdom (HD), superoxiddismutase 1 (SOD1) og TAR DNA-bindende protein 43 (TDP-43) ved amyotrofisk lateral sklerose (ALS), prionprotein (PrP) ved spongiforme encephalopatier osv. Proteinaggregater kan have indflydelse på flere cellulære veje, hvilket igen kan påvirke NRF2-niveauer og aktivitet.
Forskellige lag af regulering Styr NRF2-aktivitet tæt
Under fysiologiske forhold udviser celler lave NRF2-proteinniveauer på grund af dets hurtige omsætning. Som reaktion på forskellige stimuli akkumuleres NRF2-protein, trænger ind i kernen og øger transskriptionen af ARE-holdige gener. Derfor er styring af NRF2-proteinniveauer et nøglepunkt, der bør integrere positive og negative inputsignaler. Som vi vil diskutere yderligere, aktiveres NRF2 af forskellige overlappende mekanismer for at orkestrere en hurtig og effektiv reaktion, men på den anden side kunne NRF2 hæmmes, sandsynligvis i en anden fase, for at slukke for dens respons.
Fra det klassiske synspunkt er aktivering af NRF2 blevet betragtet som en konsekvens af den cellulære respons på oxiderende eller elektrofile forbindelser. I denne henseende spiller den ubiquitin E3-ligaseadapter Kelch-lignende ECH-associeret protein 1 (KEAP1) en afgørende rolle. Molekylære detaljer vil blive behandlet yderligere i afsnit 4.1. Kort sagt fungerer KEAP1 som en redoxsensor på grund af kritiske cysteinrester, der fører til NRF2 ubiquitinering og proteasomal nedbrydning. Ud over denne klassiske modulering er NRF2 dybt reguleret af signalbegivenheder. Faktisk har forskellige kinaser vist sig at phosphorylere og regulere NRF2. For eksempel kan NRF2 phosphoryleres af mitogenaktiverede proteinkinaser (MAPK'er), selvom dets bidrag til NRF2-aktivitet forbliver uklart [8], [9], [10], [11]. PKA-kinase såvel som nogle PKC-isozymer [12], CK2 [13] eller Fyn [14] phosphorylerer NRF2, hvilket modificerer dets stabilitet. Tidligere arbejde fra vores gruppe rapporterede, at glykogensyntase kinse-3? (GSK-3?) hæmmer NRF2 ved nuklear udelukkelse og proteasomal nedbrydning [15], [25], [26], [27], [28], [29], [30]. De molekylære detaljer vil blive diskuteret i afsnit 4.1. Endvidere er NRF2 underkastet andre former for regulering. For eksempel øger NRF2-acetylering med CBP/p300 dens aktivitet [17], mens den hæmmes af miR153, miR27a, miR142-5p og miR144 [16], eller ved methylering af cytosin-guanin (CG) øer i NRF2-promotoren [18].
Indvirkning af proteinaggregater på NRF2-reguleringsmekanismer
I dette afsnit vil vi fokusere på, hvordan akkumulering af fejlfoldet protein kan påvirke NRF2-aktivitet, hvilket giver nogle af de veje, der er nævnt ovenfor som illustrative eksempler. For det første skal vi overveje, at proteinakkumulering har været tæt forbundet med oxidativ skade. Faktisk inducerer fejlfoldet proteinakkumulering og aggregering unormal produktion af reaktive oxygenarter (ROS) fra mitokondrier og andre kilder [19]. Som nævnt ovenfor vil ROS modificere redox-følsomme cysteiner af KEAP1, hvilket fører til frigivelse, stabilisering og nuklear lokalisering af NRF2.
Med hensyn til proteinopatier er et eksempel på dysregulerede signalbegivenheder, der kan påvirke NRF2, givet af hyperaktiveringen af GSK-3? i AD. GSK-3?, også kendt som TAU-kinase, deltager i phosphoryleringen af dette mikrotubuli-associerede protein, hvilket resulterer i dets aggregering, dannelse af neurofibrillære sammenfiltringer og afbrydelse af aksonal transport (gennemgået i [20]). På den anden side GSK-3? reducerer NRF2-niveauer og aktivitet dramatisk som nævnt ovenfor. Selvom det ikke er bredt accepteret, foreslår amyloidkaskaden, at giftig A? oligomerer øger GSK-3? aktivitet sammen med TAU hyperfosforylering og neurondød [21], [22]. Der er forskellige modeller til at forklare, hvordan A? favoriserer GSK3-? aktivitet. For eksempel A? binder til insulinreceptoren og hæmmer PI3K og AKT signalveje, som er afgørende for at opretholde GSK-3? inaktiveret ved phosphorylering ved dens N-terminale Ser9-rest [23]. På den anden side, ekstracellulær A? interagerer med Frizzled-receptorer, blokerer WNT-signalering [24] og resulterer igen i frigivelse af aktivt GSK-3?. Sammenfattende, A? akkumulering fører til unormal hyperaktivering af GSK-3?, hvilket forringer et passende NRF2-respons.
Som diskuteret i det følgende afsnit fører fejlfoldede proteiner til aktivering af PERK og MAPK'er, som igen opregulerer NRF2 [31], [8], [9], [10], [11]. Desuden er dysreguleret CBP/p300-aktivitet blevet rapporteret i adskillige proteinopatier [32], og et globalt fald i DNA-methylering i AD-hjerner blev også vist [33], hvilket giver grundlag for at udforske relevansen af disse fund i NRF2-regulering.
Vi og andre har i obduktioner af PD- og AD-patienter observeret en stigning i NRF2-proteinniveauer og nogle af dets mål, såsom hæmoxygenase 1 (HMOX1), NADPH quinonoxidase 1 (NQO1), p62 osv., både ved immunoblot og ved immunhistokemi [34], [35], [36], [37], [38], [39]. Opreguleringen af NRF2 i disse sygdomme tolkes som et mislykket forsøg fra den syge hjerne på at genvinde homøostatiske værdier. En anden undersøgelse indikerede imidlertid, at NRF2 overvejende er lokaliseret i cytoplasmaet af AD hippocampale neuroner, hvilket tyder på reduceret NRF2 transkriptionel aktivitet i hjernen [40]. Det er tænkeligt, at forskellen mellem disse observationer er relateret til ændringer i de faktorer, der styrer NRF2 langs de progressive stadier af neurodegeneration.
Tre hovedsystemer bidrager til proteostase, nemlig det udfoldede proteinrespons (UPR), ubiquitin-proteasomsystemet (UPS) og autofagi. Dernæst præsenterer vi beviser for at forestille sig NRF2 som en hub, der forbinder nødsignaler aktiveret af proteinaggregater med proteinderivatmaskineriet.
NRF2 deltager i Unfolded Protein Response (UPR)
NRF2-aktivering som svar på UPR
Oxidativ proteinfoldning i ER drives af en række forskellige veje, hvoraf den mest konserverede involverer proteinet disulfid-isomerase (PDI) og sulfhydryloxidase endoplasmatisk oxidoreductin 1 (ERO1? og ERO1? i pattedyr) som disulfiddonor. Kort fortalt katalyserer PDI dannelsen og nedbrydningen af disulfidbindinger mellem cysteinrester i proteiner, når de folder, på grund af reduktionen og oxidationen af dets egne cysteinaminosyrer. PDI genanvendes ved virkningen af husholdningsenzymet ERO1, som genindfører disulfidbindinger i PDI [41]. Molekylær oxygen er den terminale elektronacceptor af ERO1, som genererer støkiometriske mængder af hydrogenperoxid for hver disulfidbinding, der produceres [42]. Peroxidaser (PRX4) og glutathionperoxidaser (GPX7 og GPX8) er nøgleenzymer til at reducere hydrogenperoxid i ER. Når dette oxidoreduktive system ikke fungerer korrekt, opstår der unormal akkumulering af fejlfoldede proteiner i ER, og et sæt signaler kaldet det udfoldede proteinrespons (UPR) overføres til cytoplasmaet og kernen for at genetablere ER-homeostasen [43]. Tre membranassocierede proteiner er blevet identificeret til at føle ER-stress i eukaryoter: aktiverende transkriptionsfaktor 6 (ATF6), pancreas ER eIF2? kinase (PERK, også dobbeltstrenget RNA-aktiveret proteinkinase-lignende ER-kinase) og inositol-krævende kinase1 (IRE1). Det luminale domæne af hver sensor er bundet til en 78 kDa chaperon kaldet glucose-reguleret protein (GRP78/BIP). BIP dissocierer ved ER-stress for at binde udfoldede proteiner, hvilket fører til aktivering af de tre sensorer [44].
NRF2 og dets homolog NRF1, også relateret til antioxidantresponset, deltager i transduktionen af UPR til kernen. I tilfælde af NRF1 er dette protein placeret ved ER-membranen og gennemgår nuklear translokation ved deglycosylering eller spaltning. Derefter fører UPR-aktivering til behandling af NRF1 og nuklear akkumulering af det resulterende fragment i det nukleare rum. Imidlertid er evnen til at transaktivere ARE-holdige gener af dette NRF1-fragment stadig under diskussion [45].
Glover-Cutter og kolleger viste aktivering af NRF2-ortologen fra C. elegans, SKN-1, med forskellige ER-stressorer. Øget SKN-1-ekspression var afhængig af forskellige UPR-mediatorer, inklusive IRE1- eller PERK-ormeortologer [46]. I PERK-mangelfulde celler fører nedsat proteinsyntese til akkumulering af endogene peroxider og efterfølgende apoptose [47]. Effektoren brugt af PERK til at beskytte ER mod disse peroxider kan være NRF2, da det er blevet rapporteret, at PERK phosphorylerer NRF2 ved Ser40 og dermed forhindrer dets nedbrydning af KEAP1 [31]. Induktionen af ASK1 vil sandsynligvis også spille en rolle i denne rute gennem den TRAF2-medierede kinasevirkning af IRE1 [48]. Selvom MAPKs rolle i reguleringen af NRF2 stadig er kontroversiel, blev det for nylig foreslået, at IRE1-TRAF2-ASK1-JNK-vejen kunne aktivere NRF2 [49] (fig. 1). Interessant nok, i C. elegans og humane celler, tyder nye beviser på, at cysteinsulfenylering af IRE1 kinase ved dets aktiveringsløkke hæmmer IRE1-medieret UPR og initierer et p38 antioxidantrespons drevet af NRF2. Dataene tyder på, at IRE1 har en gammel funktion som en cytoplasmatisk vagtpost, der aktiverer p38 og NRF2 [50].
Figur 1 Regulering af NRF2 af UPR. Akkumulering af udfoldede eller fejlfoldede proteiner inde i det endoplasmatiske retikulum kan initiere det udfoldede proteinrespons (UPR). Først frigives chaperonen BIP fra det intraluminale domæne af ER-sensorerne IRE1 og PERK for at binde udfoldede/fejlfoldede proteiner. Dette muliggør dimerisering og trans-auto-phosphorylering af deres cytosoliske domæner. PERK-aktivering resulterer i direkte NRF2-phosphorylering ved Ser40, hvilket fører til NRF2-translokation til kernen og aktivering af målgener. IRE1-aktivering inducerer rekruttering af TRAF2 efterfulgt af ASK1- og JNK-phosphorylering og aktivering. Da JNK er blevet rapporteret at phosphorylere og aktivere NRF2, er det rimeligt at tro, at IRE1-aktivering ville føre til øget NRF2-aktivitet.
Mange undersøgelser af induktionen af UPR er blevet udført med inhibitoren af proteinglycosylering tunicamycin. NRF2 ser ud til at være essentiel for forebyggelse af tunicamycin-induceret apoptotisk celledød [31], og dens aktivering under disse forhold er drevet af den autofagiske nedbrydning af KEAP1 [51]. Følgelig øgede shRNA-medieret silencing af NRF2-ekspression i βTC-6-celler, en murin insulinom β-cellelinje, signifikant tunicamycin-induceret cytotoksicitet og førte til en stigning i ekspressionen af den pro-apoptotiske ER-stressmarkør CHOP10. På den anden side reducerede NRF2-aktivering med 1,2-dithiole-3-thion (D3T) tunicamycin cytotoksicitet og svækkede ekspressionen af CHOP10 og PERK [52]. Interessant nok øgede olfaktoriske neuroner underkastet systemisk anvendelse af tunicamycin NRF2 parallelt med andre UPR-medlemmer såsom CHOP, BIP, XBP1 [53]. Disse resultater er blevet udvidet til in vivo undersøgelser, da lateral ventrikulær infusion af tunicamycin i rotter inducerede ekspression af PERK og NRF2 i hippocampus ledsaget af signifikante kognitive underskud, øget TAU-phosphorylering og A42-aflejringer [54].
NRF2 opregulerer nøglegener til vedligeholdelse af ER-fysiologien
ER-lumen har brug for en rigelig forsyning af GSH fra cytosolen for at opretholde disulfidkemi. NRF2 modulerer vigtige enzymer af GSH-metabolismen i hjernen, såsom cystin/glutamattransport, β-glutamat cysteinsyntetase (β-GS), glutamat-cysteinligase katalytiske og modulatorunderenheder (GCLC og GCLM), glutathionreduktase (GR) og glutathionperoxidase (GPX) (gennemgået i [55]). Relevansen af NRF2 i vedligeholdelsen af GSH i ER understøttes af den opdagelse, at farmakologisk eller genetisk aktivering af NRF2 resulterer i øget GSH-syntese via GCLC/GCLM, mens inhibering af ekspressionen af disse enzymer ved NRF2-knockdown forårsagede en ophobning af beskadigede proteiner i ER, der fører til UPR-aktivering [56].
I C. elegans flere komponenter af UPR-målgenerne reguleret af SKN-1, herunder Ire1, Xbp1 og Atf6. Selvom NRF2 opregulerer ekspressionen af adskillige peroxidase (PRX) og glutathionperoxidase (GPX) gener hos pattedyr (gennemgået i [57]), er kun GPX8 et bona fide ER-lokaliseret enzym, der huser KDEL-genfindingssignalet [58]. Tab af GPX8 forårsager UPR-aktivering, lækage af ERO1a-afledt hydrogenperoxid til cytosolen og celledød. Hydrogenperoxid afledt af ERO1? aktivitet kan ikke diffundere fra ER til cytosolen på grund af den samordnede virkning af GPX8 og PRX4 [59]. I denne henseende afslørede en analyse af antioxidantforsvarsvej-genekspressionsarrayet ved hjælp af RNA fra vildtype- og NRF2-nul musevæv, at ekspressionen af GPX8 var nedreguleret i fravær af NRF2 [60]. I overensstemmelse med dette viser transkriptomanalyse fra patientprøver, der lider af myeloproliferative neoplasmer, polycytæmi eller myelofibrose, sygdomme også associeret med oxidativt stress og lavgradig kronisk inflammation, lavere ekspressionsniveauer af både NRF2 og GPX8 sammenlignet med kontrolpersoner [61]. Der er endnu ikke undersøgelser, der specifikt involverer GPX8 i beskyttelse af menneskelig hjerne, men en transkriptomanalyse i mus indikerer en kompenserende GPX8-stigning som respons på Parkinson-toksinet MPTP [62].
Indvirkning af NRF2 på UPR-dysreguleringen i neurodegenerative sygdomme
Fejlfunktion af PDI-enzymer og kronisk aktivering af UPR kan efterfølgende initiere eller accelerere neurodegeneration. Sygdomsramte neuroner, dyremodeller af neurodegenerativ sygdom såvel som post-mortem humant væv viste opregulering af adskillige UPR-markører i de fleste af disse lidelser. Ændringen af PDI/UPR-vejen i neurodegenerative sygdomme er blevet pænt gennemgået i [63], men de følgende højdepunkter fra hjernens post-mortem prøver bør overvejes. PDI-niveauer er øget i tangle-bærende neuroner og i Lewy Bodies af henholdsvis AD- og PD-patienter [64], [65]. PDI og ERP57 er opreguleret i CSF fra ALS-patienter og i hjerner fra CJD-personer [66], [67], [68]. BIP, PERK, IRE1 og ATF6 er forhøjede i prøver fra patienter med AD, PD eller ALS [69], [70], [71], [67]. BIP, CHOP og XBP1 er forhøjet i post-mortem hjerneprøver fra HD [72], [73]. Desuden blev opregulering af ERP57, GRP94 og BIP fundet i cortexvæv fra CJD-patienter [74]. Alt i alt afslører disse beviser, at akkumuleringen af fejlfoldede proteiner i hjernens parenkym fører til en skadelig og kronisk aktivering af UPR. Interessant nok er der en nylig undersøgelse, der forbinder aktivering af NRF2 af PERK i begyndelsen af AD. I denne undersøgelse analyserede forfatterne, om oxidativ stress medierede ændringer i NRF2 og UPR kan udgøre tidlige begivenheder i AD patogenese ved at bruge humane perifere blodceller og en AD transgen musemodel på forskellige sygdomsstadier. Øget oxidativt stress og øget pSer40-NRF2 blev observeret i humane perifere mononukleære blodceller isoleret fra individer med mild kognitiv svækkelse. Desuden rapporterede de nedsat ER-calciumhomeostase og opregulerede ER-stressmarkører i disse celler fra individer med mild kognitiv svækkelse og mild AD [75].
Gensidig regulering af NRF2 og Ubiquitin Proteasome�System (UPS)
UPS modulerer NRF2-proteinniveauer
UPS'en deltager i nedbrydningen af beskadigede eller fejlfoldede proteiner og kontrollerer niveauerne af vigtige regulatoriske molekyler i cytosolen og kernen. Den centrale kerne i dette system er et stort multisubunit-enzym, der indeholder et proteolytisk aktivt kompleks ved navn 20S. 20S-kerneproteasomet nedbryder ufoldede proteiner, men binding til forskellige regulatoriske proteinkomplekser ændrer dets substratspecificitet og aktivitet. For eksempel udgør tilføjelsen af en eller to 19S regulatoriske underenheder til 20S-kernen 26S-proteasomet og ændrer dets specificitet mod native foldede proteiner [76], [77]. Proteasomal nedbrydning kræver kovalent binding af ubiquitin. Konjugering af ubiquitin forløber via en tre-trins kaskademekanisme. For det første aktiverer det ubiquitinaktiverende enzym E1 ubiquitin i en ATP-krævende reaktion. Derefter overfører et E2-enzym (ubiquitin-bærerprotein eller ubiquitin-konjugerende enzym) det aktiverede ubiquitin fra E1 til substratet, der er specifikt bundet til et medlem af ubiquitin-proteinligasefamilien, kaldet E3. Selvom den nøjagtige skæbne for det ubiquitinerede protein vil afhænge af arten af ubiquitinkæden, resulterer denne proces generelt i nedbrydningen af 26S-proteasomet [78].
E3-ligase KEAP1 er den bedst kendte hæmmer af NRF2. Mekanismen for KEAP1-regulering forklarer elegant, hvordan NRF2-niveauer tilpasser sig oxidantudsving. Under basale forhold gribes nysyntetiseret NRF2 af homodimeren KEAP1, som binder et NRF2-molekyle ved to aminosyresekvenser med lav (aspartat, leucin, glycin; DLG) og høj (glutamat, threonin, glycin, glutamat; ETGE) affinitet. Interaktionen med KEAP1 hjælper med at præsentere NRF2 til CULLIN3/RBX1-proteinkomplekset, hvilket resulterer i dets ubiquitinering og efterfølgende proteasomal nedbrydning. Dog hindrer redoxmodifikation af KEAP1 præsentationen af NRF2 til UPS'en repræsenteret af CULLIN3/RBX1. Som et resultat undslipper nysyntetiseret NRF2 KEAP1-afhængig nedbrydning, akkumuleres i kernen og aktiverer ARE-holdige gener [79], [80], [81], [82].
E3-ligaseadapteren a-TrCP er også en homodimer, der deltager i signalbegivenhederne relateret til phosphoryleringen af NRF2 af GSK-3a. Denne kinase phosphorylerer specifikke serinrester af NRF2 (aspartat, serin, glycin, isoleucin serin; DSGIS) for at skabe et nedbrydningsdomæne, der derefter genkendes af β-TrCP og mærkes til proteasomnedbrydning af et CULLIN1/RBX1-kompleks. Identifikationen af de specifikke aminosyrer, der phosphoryleres af GSK-3? i denne degron blev udført ved en kombination af stedrettet mutagenese af Neh6-domænet, 2D-gelelektroforese [15], [26] og massespektroskopi [83]. Følgelig hæmning af GSK-3? med meget selektive lægemidler eller siRNA'er mod GSK-3 isoformer resulterede i en stigning i NRF2-proteinniveauer. Lignende resultater blev fundet med siRNA'er mod a-TrCP isoform 1 og 2. Stabilisering af NRF2 efter GSK-3? hæmning forekom i KEAP1-deficiente museembryofibroblaster og i en ektopisk udtrykt NRF2-deletionsmutant, der mangler de kritiske ETGE-rester for højaffinitetsbinding til KEAP1, hvilket yderligere viser en KEAP1-uafhængig regulering.
I forbindelse med neurodegenerative sygdomme kan vi forestille os moduleringen af NRF2 af UPS på to forskellige måder. På den ene side ville KEAP1-systemet registrere redox-ubalance afledt af fejlfoldet proteinakkumulering, mens GSK-3/a-TrCP-aksen ville fungere som en aktiv deltager i signaltransduktion ændret ved tab af proteostase (fig. 2).
Figur 2 UPS'en styrer nøje NRF2-niveauerne. Under homøostatiske forhold opretholdes lave NRF2-niveauer af virkningen af E3-ligaseadapterne KEAP1 og β-TrCP. Til venstre binder NRF2 til Kelch-domænerne af en KEAP1-homodimer gennem et lavt (DLG) og et højt (ETGE) affinitetsmotiv. Gennem sit BTB-domæne binder KEAP1 sig samtidigt til et CULLIN3/RBX1-kompleks, hvilket muliggør NRF2-ubiquitinering og nedbrydning af 26S-proteasomet. Desuden GSK-3? phosphorylerer Ser335- og Ser338-rester af NRF2 for at skabe et nedbrydningsdomæne (DpSGIpSL), der derefter genkendes af ubiquitin-ligaseadapteren β-TrCP og mærkes til proteasomnedbrydning af et CULLIN3/RBX1-kompleks. Til højre, efter eksponering for reaktive oxygenarter eller elektrofiler modificeres kritiske Cys-rester i KEAP1, hvilket gør KEAP1 ude af stand til at interagere effektivt med NRF2 eller CULLIN3/RBX1, og derefter øger denne transkriptionsfaktor dens halveringstid og transkriptionelle aktivitet mod ARE-gener. Signaleringsveje, der resulterer i inhibering af GSK-3?, sådan AKT-phosphorylering ved Ser9, resulterer i NRF2-forringet nedbrydning af proteasomet, akkumulering og induktion af målgener.
NRF2 Øger UPS-aktivitet gennem transkriptionel kontrol af proteasomunderenheder
NRF2 opregulerer ekspressionen af flere proteasomunderenheder og beskytter således cellen mod akkumulering af toksiske proteiner. Tyve proteasom- og ubiquitinationsrelaterede gener ser ud til at være reguleret af NRF2 ifølge en bred mikroarray-analyse fra lever-RNA, der blev sat op med NRF2-induceren D3T [84]. I en posterior undersøgelse påviste de samme forfattere, at ekspressionen af de fleste underenheder af 26S-proteasomet blev forbedret op til tre gange i leveren fra mus behandlet med D3T. Underenhedsproteinniveauer og proteasomaktivitet blev koordineret øget. Der blev dog ikke set nogen induktion hos mus, hvor transkriptionsfaktoren NRF2 var forstyrret. Promotoraktivitet af PSMB5 (20S) proteasom-underenheden steg med enten NRF2-overekspression eller behandling med aktivatorer i museembryonale fibroblaster, og ARE'er blev identificeret i den proksimale promotor af PSMB5 [85]. Farmakologisk aktivering af NRF2 resulterede kun i forhøjede ekspressionsniveauer af repræsentative proteasomunderenheder (PSMA3, PSMA6, PSMB1 og PSMB5) i ikke-ældende humane fibroblaster, der indeholder funktionel NRF2 [86]. NRF2-aktivering under tilpasning til oxidativt stress resulterer i høj ekspression af PSMB1 (20S) og PA28? underenheder (eller S11, proteasomregulator) [87]. Desuden afslørede resultater fra humane embryonale stamceller, at NRF2 kontrollerer ekspressionen af proteasommodningsproteinet (POMP), en proteasomchaperon, som igen modulerer spredningen af selvfornyende humane embryonale stamceller, differentiering af tre kimlag og cellulær omprogrammering [ 88]. Alt i alt indikerer disse undersøgelser, at NRF2 opregulerer ekspressionen af nøglekomponenter i UPS'en og derfor aktivt bidrager til clearance af proteiner, der ellers ville være toksiske.
NRF2-UPS-aksen i neurodegenerative sygdomme
UPS'ens rolle i neurodegenerative sygdomme er et område med intensiv debat. Indledende undersøgelser rapporterede nedsat proteasomaktivitet i humane obduktioner af patienter ramt af adskillige neurodegenerative sygdomme. Imidlertid fandt andre undersøgelser, der anvendte in vitro og in vivo tilgange, uændret eller endda øget proteasomaktivitet (gennemgået i [89]). En mulig forklaring på denne uoverensstemmelse er, at niveauerne af UPS-komponenterne kan ændre sig under sygdomsprogression og i forskellige hjerneregioner, som det er blevet foreslået for NRF2-mål.
På trods af denne kontrovers skal det bemærkes, at opregulering af ARE-holdige proteasomgener vil forstærke UPS ved at øge clearance af toksiske proteiner i hjernen. Faktisk fører ablation af NRF1, også modulator af antioxidantresponset, i neuronale celler til nedsat proteasomaktivitet og neurodegeneration. Chromatin-immunpræcipitationseksperimenter og transkriptionel analyse viste, at PSMB6 er reguleret af NRF1. Derudover førte genekspressionsprofilering til identifikation af NRF1 som en nøgletransskriptionel regulator af proteasomgener i neuroner, hvilket tyder på, at forstyrrelser i NRF1 kan bidrage til patogenesen af neurodegenerative sygdomme [90]. Interessant nok blev NRF1 og dens lange isoform kaldet TCF11 vist at opregulere ARE-holdige proteasomgener efter proteasomhæmning i en feedback-loop for at kompensere for reduceret proteolytisk aktivitet [91], [92].
Med hensyn til NRF2 er der en sammenhæng mellem reduktion af NRF2, RPT6 (19 S) og PSMB5 (20 S) niveauer i mellemhjernen af DJ-1-deficiente mus behandlet med neurotoksinet paraquat [93]. Desuden giver den naturligt forekommende forbindelse sulforaphane (SFN) et mere robust billede af NRF2 som en afgørende modulator af UPS'en. In vitro-eksperimenter med murine neuroblastom Neuro2A-celler viste en øget ekspression af proteasomets katalytiske underenheder såvel som dets peptidaseaktiviteter som respons på SFN. Dette lægemiddel beskyttede celler mod hydrogenperoxid-medieret cytotoksicitet og proteinoxidation på en måde afhængig af proteasomfunktionen [94]. Derudover brugte Liu og kolleger en reportermus til at overvåge UPS-aktiviteten som reaktion på SFN i hjernen. Disse mus udtrykker allestedsnærværende det grønne fluorescensprotein (GFP) fusioneret til et konstitutivt nedbrydningssignal, der fremmer dets hurtige nedbrydning af UPS (GFPu). I cerebral cortex reducerede SFN niveauet af GFPu med en parallel stigning i chymotrypsin-lignende (PSMB5), caspase-lignende (PSMB2) og trypsin-lignende (PSMB1) aktiviteter af 20S-proteasomet. Derudover afslørede behandling af Huntington-afledte celler med SFN, at NRF2-aktivering forbedrede mHtt-nedbrydning og reducerede mHtt-cytotoksicitet [95]. Den vigtigste mekanisme for SFN-handling er gennem induktion af NRF2 [96]. Det specifikke bidrag fra NRF2 bør behandles ved at anvende NRF2-nul-systemer i yderligere undersøgelser.
Funktionel forbindelse mellem NRF2 og makroautofagi
NRF2-proteinniveauer moduleres af Adapter Protein P62
Autofagi refererer til nedbrydningen af cytosoliske komponenter inde i lysosomer. Denne proces bruges til at fjerne langlivede og fejlfoldede proteiner samt beskadigede organeller. En direkte forbindelse mellem NRF2 og autofagi blev først observeret i forbindelse med adapterproteinet p62, også kaldet SQSTM1 [97], [98], [99], [100], [101]. Dette protein transporterer ubiquitinerede proteiner til de proteasomale og lysosomale nedbrydningsmaskinerier og sekvestrerer beskadigede proteiner i aggregater før deres nedbrydning. P62 præsenterer et ubiquitin-associeret (UBA) domæne, til binding til ubiquitinerede proteiner, og en LC3-interagerende region (LIR) til integration med den autophagosomale membran gennem autophagy-receptoren LC3.
Selvom den p62-medierede induktion af NRF2 og dets målgener først blev rapporteret i 2007 [102], blev den molekylære mekanisme ikke fuldt ud forstået før opdagelsen af dens interaktion med KEAP1 [103], [98], [99], [100 ], [101]. Komatsu og kolleger identificerede en KEAP1 interagerende region (KIR) i p62, der bandt KEAP1 i den samme grundlæggende overfladelomme som NRF2 og med en bindingsaffinitet svarende til ETGE-motivet i NRF2, hvilket tyder på konkurrence mellem p62 og NRF2. Fosforyleringen af Ser351 i KIR-motivet i p62 (349-DPSTGE-354) blev vist at øge dens affinitet for KEAP1, konkurrere med NRF2-binding og tillade dens akkumulering og transkriptionel aktivering af dens målgener [98], [99]. Faktisk førte p62-overekspression til reduceret NRF2-ubiquitinering og deraf følgende stabilisering samt induktion af dets målgener [104]. Nogle kinaser er blevet foreslået at deltage i p62-phosphorylering. Pattedyrsmålet for rapamycinkompleks 1 (mTORC1) kan være impliceret, da behandling med mTOR-hæmmeren rapamycin undertrykte phosphoryleringen af p62 og nedreguleringen af KEAP1 ved arsenitbehandling. For nylig blev det påvist, at TGF-a-aktiveret kinase 1 (TAK1) også kunne phosphorylere p62, hvilket øgede KEAP1-nedbrydning og NRF2-opregulering. Forfatterne af denne undersøgelse foreslår, at dette er en måde at regulere cellulær redoxtase under steady-state forhold, da TAK1-mangel opregulerer ROS i fravær af nogen eksogen oxidant i forskellige musevæv parallelt med en reduktion i NRF2-proteinniveauer [105 ].
En p62-konstruktion, der manglede UBA-domænet, var stadig i stand til at binde KEAP1, hvilket antyder, at interaktionen ikke afhang af ubiquitineret KEAP1 [101]. Imidlertid indeholder p62-homologen i Drosophila melanogaster, kaldet Ref(2), ikke et KIR-motiv og interagerer ikke direkte med DmKEAP1, selvom det kan binde til ubiquitineret DmKEAP1 gennem UBA-domænet. Desuden kan DmKEAP1 direkte interagere med Atg8 (homolog til pattedyr LC3). KEAP1-mangel resulterer i Atg8 og autofagi-induktion afhængig af NRF2-ortologen CncC og uafhængig af TFEB/MITF [106]. Forholdet mellem NRF2 og autofagi ser dog ud til at være bevaret, hvilket understreger dens funktionelle relevans.
Induktionen af NRF2 af p62 er resultatet af både konkurrencen om at binde KEAP1 og nedbrydning af KEAP1 i lysosomet. Silencing af p62 med siRNA fordoblede KEAP1 halveringstid parallelt med et fald i NRF2 og dets målgener [101]. I overensstemmelse hermed viste ablation af p62-ekspression øgede niveauer af KEAP1 sammenlignet med vildtypemus. Meget relevant, stigningen i KEAP1-niveauer blev ikke påvirket af proteasomhæmmere, men blev reduceret under sult-inducerende autofagi [107]. Faktisk er KEAP1 til stede i pattedyrsceller i autofagiske vesikler dekoreret med p62 og LC3 [99], [100], [103]. Alle disse data tyder på, at KEAP1 er et substrat for makroautofagi-maskineriet, men dette spørgsmål bør analyseres mere detaljeret på grund af eksistensen af nogle kontroversielle resultater. KEAP1-proteinniveauer blev øget i Atg7-nullmus, en nøgleeffektor af makroautofagi [107], men farmakologisk hæmning af makroautofagi med torin1, E64/pepstatin eller bafilomycin kunne ikke akkumulere KEAP1 [107], [100]. Samlet set tyder disse resultater på, at øgede p62-niveauer binder KEAP1 til autofagiske vakuoler og sandsynligvis resulterer disse i KEAP1 autofagisk nedbrydning, der tillader NRF2-aktivering (fig. 3). To forskellige undersøgelser rapporterede, at sulfinsyrereduktaserne SESTRIN spiller en vigtig rolle i denne sammenhæng. SESTRIN 2 interagerer med p62, KEAP1 og RBX1 og letter p62-afhængig nedbrydning af KEAP1 og NRF2 aktivering af målgener [108]. En anden undersøgelse viste, at SESTRIN 2 interagerede med ULK1 og p62, hvilket fremmer phosphorylering af p62 ved Ser403, hvilket lettede nedbrydning af lastproteiner inklusive KEAP1 [109].
Figur 3 NRF2-niveauer reguleres af adapterproteinet p62. Fosforyleringen af Ser 351 i KIR-motivet af p62 (349-DPSTGE-354) med mTORC1, TAK1 eller andre kinaser resulterer i øget affinitet for binding til KEAP1 på grund af lighed med ETGE-motivet i NRF2. Som en konsekvens fortrænger phosphoryleret p62 NRF2 og binder KEAP1. LIR-motivet i p62 muliggør interaktion med LC3 i den autophagosomale membran, således at p62-KEAP1-komplekset til sidst nedbrydes i lysosomet. Som en konsekvens er NRF2 i stand til at akkumulere, translokere til kernen og øge transskriptionen af ARE-holdige gener, herunder p62. Denne reguleringsmekanisme giver et holdbart NRF2-respons, da KEAP1 skal syntetiseres for nylig for at hæmme NRF2-aktivitet.
Modulation af makroautofagi gener ved NRF2
NRF2 regulerer ekspressionen af relevante gener for makroautofagi såvel som det gør for UPR og UPS. Det første bevis kom fra undersøgelser, hvor p62-ekspression blev vist at blive induceret ved eksponering for elektrofiler, ROS og nitrogenoxid [110], [111], [112]. Mekanismen for induktion blev beskrevet nogle år senere med den opdagelse, at p62 indeholder en funktionel ARE i sin genpromotor [99]. I en nylig undersøgelse blev flere andre funktionelle ARE'er fundet og valideret efter bioinformatikanalyse og ChIP-assays. Desuden udviste museembryonale fibroblaster og kortikale neuroner fra Nrf2-knockout-mus reduceret p62-ekspression, som kunne reddes med et NRF2-udtrykkende lentivirus. På samme måde reducerede NRF2-mangel p62-niveauer i skadede neuroner fra mus hippocampus [36]. Derfor er det blevet foreslået, at NRF2-aktivering øger p62-niveauer, hvilket resulterer i KEAP1-nedbrydning og favoriserer yderligere NRF2-stabilisering i en positiv feedback-loop. Denne ikke-kanoniske mekanisme for NRF2-induktion kræver ændringer i genekspression og kan være et relevant svar på langvarig cellulær stress.
Lastgenkendelsesproteinet NDP52 blev vist at være transkriptionelt reguleret af NRF2. NDP52 virker på samme måde som p62, genkender ubiquitinerede proteiner og interagerer med LC3 gennem et LIR-domæne, så last nedbrydes i lysosomer. Fem formodede ARE'er blev fundet i Ndp52-promotor-DNA-sekvensen. Tre af dem blev identificeret med forskellige mutantkonstruktioner og ChIP-assays som uundværlige for NRF2-medieret Ndp52-transkription [113]. Det skal bemærkes, at Ndp52 mRNA-niveauer blev reduceret i hippocampus af Nrf2-knockout-mus. En af disse sekvenser blev også valideret i en uafhængig undersøgelse som en NRF2-reguleret ARE [36].
Imidlertid er NRF2's rolle i moduleringen af autofagi ikke begrænset til induktionen af disse to cargo-genkendelsesproteiner. For at få dybere indsigt i NRF2's rolle i moduleringen af yderligere autofagi-relaterede gener screenede vores gruppe kromatinimmunpræcipitationsdatabasen ENCODE for to proteiner, MAFK og BACH1, som binder de NRF2-regulerede ARE'er. Ved at bruge et script genereret fra JASPAR's konsensus ARE-sekvens, identificerede vi flere formodede ARE'er i mange autofaggener. Tolv af disse sekvenser blev valideret som NRF2-regulerede ARE'er i ni autofaggener, hvis ekspression blev formindsket i museembryofibroblaster fra Nrf2-knockout-mus, men kunne genoprettes af et NRF2-udtrykkende lentivirus. Vores undersøgelse viste, at NRF2 aktiverer ekspressionen af nogle gener involveret i forskellige trin af den autofagiske proces, herunder autofagi-initiering (ULK1), lastgenkendelse (p62 og NDP52), autophagosomdannelse (ATG4D, ATG7 og GABARAPL1), forlængelse (ATG2B og ATG5). ), og autolysosom clearance (ATG4D). Følgelig blev autophagy flux som reaktion på hydrogenperoxid svækket, når NRF2 var fraværende [36].
Relevans af NRF2-medieret makroautofagi-generekspression i neurodegenerative lidelser
Defekt autofagi har vist sig at spille en vigtig rolle i adskillige neurodegenerative sygdomme [114], og ablation af autofagi fører til neurodegeneration hos mus [115], [116]. Atg7-knockout-mus afslørede, at autophagy-mangel resulterer i p62-akkumulering i ubiquitin-positive inklusionslegemer. KEAP1 blev sekvestreret i disse inklusionslegemer, hvilket førte til NRF2-stabilisering og induktion af målgener [103]. Det er vigtigt, at overdreven akkumulering af p62 sammen med ubiquitinerede proteiner er blevet identificeret i neurodegenerative sygdomme, herunder AD, PD og ALS [117]. Faktisk udtrykte neuroner, der udtrykte høje niveauer af APP eller TAU hos AD-patienter, også p62 og nuklear NRF2, hvilket tyder på deres forsøg på at nedbryde intraneuronale aggregater gennem autofagi [36].
NRF2-mangel forværrer proteinaggregering i forbindelse med AD. Faktisk findes øgede niveauer af phosphoryleret og sarkosyl-uopløselig TAU i Nrf2-knockout-mus, selvom ingen forskel i kinase- eller phosphatase-aktiviteter kunne påvises sammenlignet med vildtype-baggrunden [113]. Det er vigtigt, at NDP52 blev vist at co-lokalisere med TAU i murine neuroner, og direkte interaktion mellem phospho-TAU og NDP52 blev vist ved co-immunpræcipitationseksperimenter både i mus og AD-prøver, hvilket peger på dets rolle i TAU-nedbrydning. Interessant nok resulterede silencing af NDP52, p62 eller NRF2 i neuroner i øget phospho-TAU [113], [118]. Desuden blev der fundet øgede intraneuronale APP-aggregater i hippocampus af APP/PS1?E9-mus, når NRF2 var fraværende. Dette korrelerede med ændrede autofagi-markører, herunder øgede phospho-mTOR/mTOR- og phospho-p70S6k/p70S6k-forhold (indikerende for autofagi-hæmning), forøgede niveauer af præ-cathepsin D og et større antal multivesikulære legemer [119]. Hos mus, der co-udtrykker human APP (V717I) og TAU (P301L), førte NRF2-mangel til øgede niveauer af total og phospho-TAU i den uopløselige fraktion og øgede intraneuronale APP-aggregater sammen med reducerede neuronale niveauer af p62, NDP52, ULK1, ATG5 og GABARAPL1. Co-lokalisering mellem adapterproteinet p62 og APP eller TAU blev reduceret i fravær af NRF2 [36]. Samlet set fremhæver disse resultater vigtigheden af NRF2 i neuronal autofagi.
Forskellige transkriptionsfaktorer virker koordineret for at modulere proteostase
Under steady state-betingelser styres proteostase via protein-protein-interaktioner og post-translationelle modifikationer, der opnår en hurtig respons. Imidlertid kræver cellulær tilpasning transkriptionel regulering af UPR, UPS og autophagy gener. I betragtning af at nerveceller kontinuerligt udsættes for lavgradige toksiske fornærmelser, herunder oxidativt og proteotoksisk stress, kan en forstærkning af proteostase induceret af transkriptionel modulering hjælpe med at forhindre hjernedegeneration.
I tilfælde af UPR vil aktiveringen af hver af de tre arme endelig resultere i transkriptionel induktion af visse gener (gennemgået i [43]). For eksempel binder et ATF6-afledt fragment (ATF6f) til ER-stress responselementer (ERSE) og inducerer ekspressionen af flere gener, herunder XBPI, BIP og CHOP. Derudover fører PERK-signalering til aktivering af transkriptionsfaktoren ATF4, som kontrollerer ekspressionen af flere UPR-relaterede gener og nogle andre, herunder NRF2-målgenerne Hmox1 og p62. Endelig resulterer IRE1-aktivering i dannelsen af en aktiv transkriptionsfaktor, splejset XBP1 (XBP1s), som kontrollerer transskriptionen af gener, der koder for proteiner involveret i proteinfoldning.
På den anden side blev NRF1 vist at være nødvendig for proteasomal genekspression i hjernen, da Nrf1-knockout-mus udviste reduceret ekspression af gener, der koder for forskellige underenheder af 20S-kernen, såvel som det regulatoriske 19S-kompleks sammen med nedsat proteasomal funktion [90 ]. Både NRF1 og NRF2 binder til ARE-sekvenser i promotorregionerne af dets målgener, hvilket tyder på, at de har overlappende transkriptionelle aktiviteter, selvom de adskiller sig i deres reguleringsmekanismer og cellulære lokalisering [120].
Transkriptionsfaktorer af Forkhead box O (FOXO)-familien styrer ekspressionen af flere autofagi-relaterede gener. I lighed med, hvad der sker med NRF2, er der flere lag af regulering af aktiviteten af FOXO-medlemmer, som kan induceres ved ernæringsmæssigt eller oxidativt stress [121]. Endelig spiller transkriptionsfaktoren TFEB, der betragtes som hovedregulatoren af lysosomal biogenese, en afgørende rolle i reguleringen af autofagi under ernæringsmæssige stressforhold. Således fører inhibering af mTORC1 til nuklear translokation af TFEB og induktion af ekspressionen af autofaggener [122].
Samlet set antyder eksistensen af forskellige transkriptionelle regulatorer af disse maskinerier også krydstale og delvist overflødige mekanismer, der kan sikre proteostase under forskellige omstændigheder. Følgelig kan NRF2 have en relevant rolle i væv, der understøtter høje niveauer af oxidativ stress. For eksempel kan oxidativ stress-induceret NRF2 fungere under næringsrige forhold for at transskriptionelt opregulere autofagi, svarende til hvad der er blevet fundet for TFEB under sultforhold. Desuden fungerer hjernen stort set under næringsrige forhold, hvilket udgør NRF2 som en relevant mekanisme til at aktivere autofagi i neuroner.
Lovende�Terapeutisk potentiale for NRF2 i proteinopatier
I de sidste par år er der sket et stort fremskridt i viden om de regulerende roller af UPR, UPS og autofagi på NRF2-aktivitet, såvel som den gensidige NRF2-medierede transskription af komponenter i disse tre systemer. Derfor kan der opstå nye terapeutiske muligheder baseret på udnyttelsen af NRF2 som en afgørende regulator af proteinclearance i neurodegenerative sygdomme.
Et centralt tilbageværende spørgsmål er imidlertid, om det vil være nyttigt eller skadeligt at øge NRF2-niveauer i hjernen. Analyse af epidemiologiske data kan give et delvist svar, da det indikerer, at NFE2L2-genet er meget polymorft, og nogle enkeltnukleotidpolymorfier fundet i dets promotorregulerende region kan give en række �fysiologiske� variationer i genekspression på populationsniveau og nogle haplotyper var forbundet med nedsat risiko og/eller forsinket indtræden af AD, PD eller ALS [123]. Desuden, som diskuteret af Hayes og kolleger [124], kan NRF2-effekten have en U-formet respons, hvilket betyder, at for lave NRF2-niveauer kan resultere i tab af cytobeskyttelse og øget modtagelighed for stressorer, mens for meget NRF2 kan forstyrre den homøostatiske balance i forhold til et reduktivt scenarie (reduktivt stress), som ville fremme proteinfejlfoldning og aggregering. Lave NRF2-niveauer i hjernen understøtter ideen om, at en let opregulering kan være tilstrækkelig til at opnå en fordel under patologiske forhold. Faktisk er den beskyttende rolle af farmakologisk NRF2-medieret aktivering af proteinclearance blevet vist i forskellige neurodegenerationscellekulturer og in vivo-modeller.
SFN er en farmakologisk NRF2-aktivator, der blev vist at inducere proteasomal og autofagi genekspression [95], [36]. Interessant nok viste Jo og kolleger, at SFN reducerede niveauerne af phosphoryleret TAU og øgede Beclin-1 og LC3-II, hvilket tyder på, at NRF2-aktivering kan lette nedbrydning af dette toksiske protein gennem autofagi [113]. Desuden blev nedbrydning af mHtt forstærket med SFN, og dette blev vendt tilbage med brugen af MG132, hvilket indikerer proteasomal nedbrydning af dette toksiske protein [95]. Autofagi-medieret nedbrydning af phospho- og uopløseligt TAU blev rapporteret med det organiske flavonoid fisetin. Denne forbindelse var i stand til at inducere autofagi ved samtidig at fremme aktiveringen og nuklear translokation af både TFEB og NRF2 sammen med nogle af dets målgener. Dette svar blev forhindret af TFEB- eller NRF2-dæmpning [125]. Bott og kolleger rapporterede gavnlige virkninger af en samtidig NRF2-, NRF1- og HSF1-aktivator på proteintoksicitet i spinal og bulbar muskelatrofi, en neurodegenerativ lidelse forårsaget af udvidelse af polyglutamin-kodende CAG-gentagelser, hvori proteinaggregater er til stede [126]. Potentialet for NRF2-aktivering til behandling af neurodegenerative lidelser er blevet demonstreret med godkendelsen af BG-12, den orale formulering af NRF2-induceren dimethylfumarat (DMF), til behandling af dissemineret sklerose [127], [128]. Succesen med DMF med autoimmune sygdomme med en stærk inflammatorisk komponent tyder på, at neurodegenerative sygdomme kan have gavn af at omplacere dette lægemiddel. I en nylig præklinisk undersøgelse af en β-synukleinopati-model af PD blev DMF vist at være neurobeskyttende, delvist på grund af dets induktion af autofagi [129]. Undersøgelser, der rapporterer gavnlige virkninger af NRF2 på neurodegeneration, men ikke fokuserer på dets effekt på proteinclearance, er endnu mere rigelige (for en omfattende gennemgang, se [7]). Dette er ret relevant, da det fremhæver de mange skadelige processer, der samtidigt kan målrettes af et enkelt hit i NRF2, herunder oxidativt stress, neuroinflammation eller mitokondriel dysfunktion. Fremtidigt arbejde vil dog være nødvendigt for at afgøre, om farmakologisk aktivering af NRF2 kan være en gyldig strategi til at lette nedbrydning af toksiske proteiner i hjernen.
Som forklaret før, forværret GSK-3? aktivitet blev rapporteret i neurodegenerative sygdomme, og det er blevet spekuleret i, at deraf følgende NRF2-reduktion kan være delvist ansvarlig for det skadelige resultat. Under disse patologiske forhold kunne GSK-3-hæmmere også samarbejde for at øge NRF2-niveauer og proteostase. De gavnlige virkninger af GSK-3-hæmmere er blevet rapporteret i forskellige modeller af neurodegeneration, og mere interessant, GSK-3-repression blev vist at reducere niveauerne af toksiske proteiner [130], [131], [132], [133]. Selvom der endnu ikke er observeret nogen direkte forbindelser mellem GSK-3-hæmning og NRF2-transkriptionel regulering af gener, der fremmer proteostase, er det rimeligt at spekulere i, at nedregulering af GSK-3-aktivitet vil resultere i øgede NRF2-niveauer, hvilket i sidste ende vil resultere i forstærkede proteostase.
Den transkriptionelle aktivitet af NRF2 såvel som den cellulære kapacitet til at opretholde proteostase falder med alderen, den vigtigste risikofaktor for udvikling af neurodegenerative sygdomme. Det er rimeligt at tro, at forstærkningen af NRF2 og følgelig proteostase i det mindste ville forsinke akkumuleringen af proteinaggregater og neurodegeneration. Faktisk fremmede behandling af humane senescent fibroblaster med 18β-glycyrrhetinsyre (18β-GA) triterpenoid NRF2-aktivering, hvilket førte til proteasominduktion og forlænget levetid. Denne undersøgelse tyder på, at farmakologisk aktivering af NRF2 er mulig selv i det sene liv [86]. Desuden viste en senere undersøgelse, at denne forbindelse medierede SKN-1 og proteasomaktivering i C.elegans med gavnlige effekter på AD progression i relevante nematodemodeller [134].
Alt taget i betragtning, synes NRF2-medieret induktion af proteostase-relaterede gener at være gavnlig i forskellige proteinopatier.
Sulforaphane og dens virkninger på kræft, dødelighed, aldring, hjerne og adfærd, hjertesygdomme og mere
Isothiocyanater er nogle af de vigtigste planteforbindelser, du kan få i din kost. I denne video gør jeg det mest omfattende tilfælde til dem, der nogensinde er blevet lavet. Kort opmærksomhed span? Spring til dit yndlingsemne ved at klikke på et af nedenstående tidspunkter. Fuld tidslinje nedenfor.
Nøglesektioner:
00: 01: 14 - Kræft og dødelighed
00: 19: 04 - Aging
00: 26: 30 - Hjerne og adfærd
00: 38: 06 - Final recap
00: 40: 27 - Dosis
Fuld tidslinje:
00: 00: 34 - Introduktion af sulforaphane, et vigtigt fokus i videoen.
00: 01: 14 - Cruciferous vegetabilsk forbrug og reduktion af allårsag dødelighed.
00: 02: 12 - Prostatacancerrisiko.
00: 02: 23 - Kræftrisiko.
00: 02: 34 - Lungekræft i rygere risiko.
00: 02: 48 - Brystkræftrisiko.
00: 03: 13 - Hypotetisk: Hvad hvis du allerede har kræft? (Interventionel)
00: 03: 35 - Plausibel mekanisme, der driver kræft- og dødelighedsassosierende data.
00: 04: 38 - Sulforaphane og kræft.
00: 05: 32 - Dyrebevis, der viser stærk virkning af broccoli-spireekstrakt på udviklingen af blæretumor hos rotter.
00: 06: 06 - Virkning af direkte tilsætning af sulforaphan hos patienter med prostatacancer.
00: 07: 09 - Bioakkumulering af isothiocyanatmetabolitter i egentligt brystvæv.
00: 08: 32 - Inhibering af brystcancerstamceller.
00: 08: 53 - Historie lektion: brassicas blev etableret som at have sundhedsegenskaber selv i det gamle Rom.
00: 09: 16 - Sulforaphans evne til at øge kræftfremkaldende udskillelse (benzen, acrolein).
00: 09: 51 - NRF2 som en genetisk switch via antioxidant responselementer.
00: 10: 10 - Hvordan NRF2 aktivering øger kræftfremkaldende udskillelse via glutathion-S-konjugater.
00: 10: 34 - Spiraler øger glutathion-S-transferase og reducerer DNA-beskadigelse.
00: 11: 20 - Broccoli-spire drikker øger benzenudskillelsen med 61%.
00: 13: 31 - Broccoli spirehomogenat øger antioxidant enzymer i den øvre luftvej.
00: 15: 45 - Cruciferous vegetabilsk forbrug og dødelighed i hjertesygdomme.
00: 16: 55 - Broccoli spire pulver forbedrer blodlipider og overordnet hjertesygdomsrisiko hos type 2 diabetikere.
00: 19: 04 - Begyndelse af aldringssektionen.
00: 19: 21 - Sulforaphane beriget kost forbedrer levetiden for biller fra 15 til 30% (under visse forhold).
00: 20: 34 - Betydningen af lav inflammation for lang levetid.
00: 22: 05 - Cruciferous grøntsager og broccoli spire pulver synes at reducere et bredt udvalg af inflammatoriske markører hos mennesker.
00: 24: 14 - Musestudier tyder på, at sulforaphan kan forbedre adaptiv immunfunktion i alderdommen.
00: 25: 18 - Sulforaphane forbedrede hårvækst i en musmodel af skaldethed. Billede på 00: 26: 10.
00: 26: 30 - Begyndelse af hjerne- og adfærdssektionen.
00: 27: 18 - Effekt af broccoli-spireekstrakt på autisme.
00: 27: 48 - Virkning af glucoraphanin på skizofreni.
00: 28: 17 - Start af depression diskussion (sandsynlig mekanisme og undersøgelser).
00: 31: 21 - Mus undersøgelse ved hjælp af 10 forskellige modeller af stress-induceret depression viser sulforaphan på samme måde som fluoxetin (prozac).
00: 32: 00 - Undersøgelse viser direkte indtagelse af glucoraphanin hos mus er tilsvarende effektiv til forebyggelse af depression fra social nederlagsspændingsmodel.
00: 33: 01 - Begyndelsen af neurodegenerationsafsnittet.
00: 33: 30 - Sulforaphane og Alzheimers sygdom.
00: 33: 44 - Sulforaphane og Parkinsons sygdom.
00: 33: 51 - Sulforaphane og Hungtington's sygdom.
00: 35: 55 - Sulforaphane og neuronal plasticitet.
00: 36: 32 - Sulforaphane forbedrer læring som model af type II diabetes hos mus.
00: 37: 19 - Sulforaphane og duchenne muskeldystrofi.
00: 37: 44 - Myostatininhibering i muskel-satellitceller (in vitro).
00: 38: 06 - Late video recap: dødelighed og kræft, DNA-beskadigelse, oxidativ stress og betændelse, udslip af benzen, kardiovaskulær sygdom, type II diabetes, hjernens virkninger (depression, autisme, skizofreni, neurodegeneration), NRF2-pathway.
00: 40: 27 - Tanker om at finde ud af en dosis broccolispirer eller sulforaphane.
00: 41: 01 - Anecdoter på spiring hjemme.
00: 43: 14 - På tilberedningstemperaturer og sulforafanaktivitet.
00: 43: 45 - Gut-bakteriekonvertering af sulforaphan fra glucoraphanin.
00: 44: 24 - Tilskud fungerer bedre, når de kombineres med aktiv myrosinase fra grøntsager.
00: 44: 56 - Madlavningsteknikker og cruciferous grøntsager.
00: 46: 06 - Isothiocyanater som goitrogener.
Nuklear faktor erythroid-afledt 2 (NF-E2)-relateret faktor 2, ellers kendt som Nrf2, er en transkriptionsfaktor, som regulerer ekspressionen af en række antioxidanter og afgiftende enzymer. Forskningsundersøgelser har også vist dens rolle i at kontrollere oxidativ stress. De fleste neurodegenerative sygdomme, såsom Alzheimers sygdom og Parkinsons sygdom, er karakteriseret ved oxidativt stress og kronisk inflammation, de almindelige mål for Nrf2 behandlingstilgange. Dr. Alex Jimenez DC, CCST Insight
Afsluttende bemærkninger
Transkriptionsfaktor NRF2 orkestrerer en proteostatisk respons ved at registrere og modulere ændringer i UPR, UPS og autofagi (fig. 4). Som følge heraf har manglen på NRF2 vist sig at forværre proteinopati, hvilket tyder på, at NRF2 er nødvendig for optimal proteinclearance. Alt sammen kan vi spekulere i, at NRF2 kan være et interessant terapeutisk mål for proteinopatier.
Figur 4 NRF2 som en hub, der forbinder proteotoksisk-afledte nødsignaler til en beskyttende transkriptionel reaktion. Akkumuleringen af udfoldede/fejlfoldede proteiner vil føre til aktivering af det udfoldede proteinrespons (UPR) i ER. Aktivering af PERK eller MAPK kan resultere i transkriptionel induktion af den ER-resident Gpx8 og adskillige enzymer, der regulerer GSH-niveauer, afgørende for at sikre korrekt proteinfoldning. Proteinaggregater hæmmer proteasomaktivitet (UPS), hvilket sandsynligvis undgår NRF2-nedbrydning. NRF2 har vist sig specifikt at modulere transkriptionen af Psma3, Psma6, Psmb1, Psmb5 og Pomp gener. Adskillige andre underenheder blev opreguleret på en NRF2-afhængig måde som svar på D3T, hvilket sandsynligvis udvidede listen over proteasomunderenheder reguleret af NRF2. Autofagi er hovedvejen for nedbrydning af proteinaggregater. Autophagy regulerer også NRF2, der forbinder denne nedbrydningsvej med NRF2 transkriptionel induktion af p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 og Gabarapl1.
Ifølge artiklen ovenfor, mens symptomerne på neurodegenerative sygdomme kan behandles gennem en række forskellige behandlingsmuligheder, har forskningsundersøgelser vist, at Nrf2-aktivering kan være en nyttig behandlingstilgang. Fordi Nrf2-aktivatorer retter sig mod brede sygdomsmekanismer, kan alle neurodegenerative sygdomme drage fordel af brugen af Nrf2-transskriptionsfaktoren. Resultaterne af Nrf2 har revolutioneret behandlingen af neurodegenerative sygdomme. Omfanget af vores information er begrænset til kiropraktik og rygsøjlesundhedsproblemer. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Yderligere emne diskussion: Relieving knæsmerter uden kirurgi
Knæsmerter er et velkendt symptom, der kan opstå på grund af en række knæskader og / eller tilstande, herunder sportsskader. Knæet er et af de mest komplekse led i den menneskelige krop, da det består af skæringspunktet mellem fire knogler, fire ledbånd, forskellige sener, to menisker og brusk. Ifølge American Academy of Family Physicians omfatter de mest almindelige årsager til knæsmerter patellar subluksation, patellar tendinitis eller jumper's knee og Osgood-Schlatters sygdom. Selvom knæsmerter mest sandsynligt forekommer hos personer over 60 år, kan knæsmerter også forekomme hos børn og unge. Knæsmerter kan behandles hjemme efter RICE-metoderne, dog kan alvorlige knæskader kræve øjeblikkelig lægehjælp, herunder kiropraktisk behandling. �
Oxidativt stress beskrives som celleskade forårsaget af frie radikaler eller ustabile molekyler, som i sidste ende kan påvirke sund funktion. Den menneskelige krop skaber frie radikaler for at neutralisere bakterier og vira, men eksterne faktorer, såsom ilt, forurening og stråling, kan ofte også producere frie radikaler. Oxidativ stress har været forbundet med adskillige sundhedsproblemer.
Oxidativ stress og andre stressfaktorer aktiverer interne beskyttelsesmekanismer, som kan hjælpe med at regulere den menneskelige krops antioxidantrespons. Nrf2 er et protein, som fornemmer niveauer af oxidativt stress og gør det muligt for cellerne at beskytte sig selv mod interne og eksterne faktorer. Nrf2 har også vist sig at hjælpe med at regulere gener involveret i produktionen af antioxidantenzymer og stress-respons gener. Formålet med artiklen nedenfor er at forklare effekter af Nrf2 i kræft.
Abstrakt
Keap1-Nrf2-vejen er den vigtigste regulator af cytobeskyttende reaktioner på oxidativt og elektrofilt stress. Selvom cellesignalveje udløst af transkriptionsfaktoren Nrf2 forhindrer kræftinitiering og -progression i normalt og præmalignt væv, giver Nrf2-aktivitet vækstfordele i fuldt maligne celler ved at øge cancerkemoresistens og øge tumorcellevækst. I denne grafiske gennemgang giver vi et overblik over Keap1-Nrf2-vejen og dens dysregulering i kræftceller. Vi opsummerer også kort konsekvenserne af konstitutiv Nrf2-aktivering i kræftceller, og hvordan dette kan udnyttes i kræftgenterapi.
Keap1-Nrf2-vejen er den vigtigste regulator af cytobeskyttende reaktioner på endogene og eksogene belastninger forårsaget af reaktive oxygenarter (ROS) og elektrofiler [1]. De vigtigste signalproteiner i vejen er transkriptionsfaktoren Nrf2 (nuklear faktor erythroid 2-relateret faktor 2), der binder sammen med små Maf-proteiner til antioxidantresponselementet (ARE) i de regulatoriske regioner af målgener, og Keap1 (Kelch ECH associerer protein 1), et repressorprotein, der binder til Nrf2 og fremmer dets nedbrydning af ubiquitin-proteasomvejen (fig. 1). Keap1 er et meget cysteinrigt protein, mus Keap1 har i alt 25 og humane 27 cysteinrester, hvoraf de fleste kan modificeres in vitro af forskellige oxidanter og elektrofiler [2]. Tre af disse rester, C151, C273 og C288, har vist sig at spille en funktionel rolle ved at ændre konformationen af Keap1, hvilket fører til nuklear translokation af Nrf2 og efterfølgende målgenekspression [3] (fig. 1). Den nøjagtige mekanisme, hvorved cysteinmodifikationer i Keap1 fører til Nrf2-aktivering, kendes ikke, men de to fremherskende, men ikke gensidigt udelukkende modeller er (1) hængsel- og låsemodellen, hvor Keap1-modifikationer i thiolrester, der findes i IVR af Keap1 forstyrre interaktionen med Nrf2, hvilket forårsager en fejljustering af lysinresterne i Nrf2, der ikke længere kan polyubiquitinyleres, og (2) modellen, hvor thiolmodifikation forårsager dissociation af Cul3 fra Keap1 [3]. I begge modeller er den inducer-modificerede og Nrf2-bundne Keap1 inaktiveret, og som følge heraf omgår nysyntetiserede Nrf2-proteiner Keap1 og translokeres ind i kernen, binder til ARE og driver ekspressionen af Nrf2-målgener såsom NAD(P)H quinonoxidoreduktase 1 (NQO1), hæmoxygenase 1 (HMOX1), glutamat-cysteinligase (GCL) og glutathion S-transferaser (GST'er) (fig. 2). Ud over modifikationer af Keap1 thioler, der resulterer i Nrf2 målgeninduktion, kan proteiner såsom p21 og p62 binde til Nrf2 eller Keap1 og derved forstyrre interaktionen mellem Nrf2 og Keap1 [1], [3] (fig. 3).
Fig. 1. Strukturer af Nrf2 og Keap1 og cysteinkoden. (A) Nrf2 består af 589 aminosyrer og har seks evolutionært højt konserverede domæner, Neh1-6. Neh1 indeholder et bZip-motiv, en basisregion � leucin lynlås (L-Zip) struktur, hvor den grundlæggende region er ansvarlig for DNA-genkendelse, og L-Zip medierer dimerisering med små Maf-proteiner. Neh6 fungerer som en degron til at mediere nedbrydning af Nrf2 i kernen. Neh4 og 5 er transaktiveringsdomæner. Neh2 indeholder ETGE- og DLG-motiver, som er nødvendige for interaktionen med Keap1, og en hydrofil region af lysinrester (7 K), som er uundværlige for den Keap1-afhængige polyubiquitinering og nedbrydning af Nrf2. (B) Keap1 består af 624 aminosyrerester og har fem domæner. De to protein-proteininteraktionsmotiver, BTB-domænet og Kelch-domænet, er adskilt af den intervenerende region (IVR). BTB-domænet sammen med den N-terminale del af IVR medierer homodimerisering af Keap1 og binding med Cullin3 (Cul3). Kelch-domænet og den C-terminale region medierer interaktionen med Neh2. (C) Nrf2 interagerer med to Keap1-molekyler gennem dets Neh2 ETGE- og DLG-motiver. Både ETGE og DLG binder til lignende steder på bunden af Keap1 Kelch-motivet. (D) Keap1 er rig på cysteinrester med 27 cysteiner i humant protein. Nogle af disse cysteiner er placeret i nærheden af basiske rester og er derfor fremragende mål for elektrofiler og oxidanter. Modifikationsmønsteret af cysteinresterne af elektrofiler er kendt som cysteinkoden. Cysteinkodehypotesen foreslår, at strukturelt forskellige Nrf2-aktiverende midler påvirker forskellige Keap1-cysteiner. Cysteinmodifikationerne fører til konformationelle ændringer i Keap1, der forstyrrer interaktionen mellem Nrf2 DLG- og Keap1 Kelch-domænerne, og dermed hæmmer polyubiquitineringen af Nrf2. Den funktionelle betydning af Cys151, Cys273 og Cys288 er blevet vist, da Cys273 og Cys288 er nødvendige for undertrykkelse af Nrf2 og Cys151 for aktivering af Nrf2 af inducere [1], [3].
Fig. 2. Nrf2-Keap1 signalvejen. (A og B) under basale forhold binder to Keap1-molekyler til Nrf2, og Nrf2 polyubiquityleres af det Cul3-baserede E3-ligasekompleks. Denne polyubiquitilation resulterer i hurtig Nrf2-nedbrydning af proteasomet. En lille del af Nrf2 undslipper det hæmmende kompleks og akkumuleres i kernen for at mediere basal ARE-afhængig genekspression og derved opretholde den cellulære homeostase. (C) Under stressforhold modificerer inducere Keap1-cysteinerne, hvilket fører til inhibering af Nrf2 ubiquitylering via dissociation af det hæmmende kompleks. (D) Ifølge hængsel- og låsemodellen fører modifikation af specifikke Keap1-cysteinrester til konformationelle ændringer i Keap1, hvilket resulterer i løsrivelsen af Nrf2 DLG-motivet fra Keap1. Ubiquitinering af Nrf2 afbrydes, men bindingen med ETGE-motivet forbliver. (E) I Keap1-Cul3-dissociationsmodellen afbrydes bindingen af Keap1 og Cul3 som reaktion på elektrofiler, hvilket fører til undslippet af Nrf2 fra ubiquitineringssystemet. I begge de foreslåede modeller er den inducer-modificerede og Nrf2-bundne Keap1 inaktiveret, og som følge heraf omgår nysyntetiserede Nrf2-proteiner Keap1 og translokerer ind i kernen, binder til Antioxidant Response Element (ARE) og driver ekspressionen af Nrf2-målet. gener såsom NQO1, HMOX1, GCL og GST'er [1], [3].
Fig. 3. Mekanismer for konstitutiv nuklear akkumulering af Nrf2 i cancer. (A) Somatiske mutationer i Nrf2 eller Keap1 forstyrrer interaktionen mellem disse to proteiner. I Nrf2 påvirker mutationer ETGE- og DLG-motiver, men i Keap1 er mutationer mere jævnt fordelt. Desuden kan onkogenaktivering, såsom KrasG12D[5], eller afbrydelse af tumorsuppressorer, såsom PTEN [11] føre til transkriptionel induktion af Nrf2 og en stigning i nuklear Nrf2. (B) Hypermethylering af Keap1-promotoren i lunge- og prostatacancer fører til reduktion af Keap1-mRNA-ekspression, hvilket øger den nukleare akkumulering af Nrf2 [6], [7]. (C) Ved familiært papillært nyrecarcinom fører tabet af fumarathydratase-enzymaktivitet til akkumulering af fumarat og yderligere til succination af Keap1-cysteinrester (2SC). Denne post-translationelle modifikation fører til afbrydelse af Keap1-Nrf2 interaktion og nuklear akkumulering af Nrf2 [8], [9]. (D) Akkumulering af forstyrrende proteiner såsom p62 og p21 kan forstyrre Nrf2-Keap1-binding og resulterer i en stigning i nuklear Nrf2. p62 binder til Keap1 og overlapper bindingslommen for Nrf2, og p21 interagerer direkte med DLG- og ETGE-motiverne af Nrf2 og konkurrerer derved med Keap1 [10].
Mekanismer for aktivering og dysregulering af Nrf2 i kræft
Selvom cytobeskyttelse leveret af Nrf2-aktivering er vigtig for cancer-kemoforebyggelse i normalt og præmalignt væv, giver Nrf2-aktivitet vækstfordele i fuldt maligne celler ved at øge cancer-kemoresistens og øge tumorcellevækst [4]. Adskillige mekanismer, hvorved Nrf2-signalvejen er konstitutivt aktiveret i forskellige cancerformer, er blevet beskrevet: (1) somatiske mutationer i Keap1 eller Keap1-bindingsdomænet af Nrf2, der forstyrrer deres interaktion; (2) epigenetisk dæmpning af Keap1-ekspression, der fører til defekt undertrykkelse af Nrf2; (3) akkumulering af forstyrrende proteiner, såsom p62, der fører til dissociation af Keap1-Nrf2-komplekset; (4) transkriptionel induktion af Nrf2 ved onkogene K-Ras, B-Raf og c-Myc; og (5) post-translationel modifikation af Keap1-cysteiner ved succinylering, der forekommer i familiært papillært nyrecarcinom på grund af tab af fumarathydratase-enzymaktivitet [3], [4], [5], [6], [7], [ 8], [9], [10] (fig. 3). Konstitutivt rigeligt Nrf2-protein forårsager øget ekspression af gener involveret i lægemiddelmetabolisme og øger derved modstanden over for kemoterapeutiske lægemidler og strålebehandling. Derudover er højt Nrf2-proteinniveau forbundet med dårlig prognose ved cancer [4]. Overaktiv Nrf2 påvirker også celleproliferation ved at dirigere glucose og glutamin mod anabolske veje, der øger purinsyntesen og påvirker pentosephosphatvejen for at fremme celleproliferation [11] (fig. 4).
Fig. 4. Den dobbelte rolle af Nrf2 i tumorgenese. Under fysiologiske forhold er lave niveauer af nuklear Nrf2 tilstrækkelige til at opretholde cellulær homeostase. Nrf2 hæmmer tumorinitiering og cancermetastaser ved at eliminere kræftfremkaldende stoffer, ROS og andre DNA-skadelige stoffer. Under tumorgenese fører akkumulering af DNA-skader til konstitutiv hyperaktivitet af Nrf2, som hjælper de autonome maligne celler til at udholde høje niveauer af endogen ROS og undgå apoptose. Vedvarende forhøjede nukleare Nrf2-niveauer aktiverer metaboliske gener ud over de cytobeskyttende gener, der bidrager til metabolisk omprogrammering og forbedret celleproliferation. Kræfter med høje Nrf2-niveauer er forbundet med dårlig prognose på grund af radio- og kemoresistens og aggressiv cancercelleproliferation. Således er Nrf2 pathway-aktivitet beskyttende i de tidlige stadier af tumorigenese, men skadelig i de senere stadier. Til forebyggelse af kræft er det derfor fortsat en vigtig tilgang at øge Nrf2-aktivitet, hvorimod Nrf2-hæmning er ønskelig til behandling af kræft [4], [11].
I betragtning af at høj Nrf2-aktivitet almindeligvis forekommer i kræftceller med uønskede resultater, er der behov for terapier til at hæmme Nrf2. På grund af strukturel lighed med nogle andre bZip-familiemedlemmer er udviklingen af specifikke Nrf2-hæmmere desværre en udfordrende opgave, og kun få undersøgelser af Nrf2-hæmning er blevet offentliggjort til dato. Ved at screene naturlige produkter har Ren et al. [12] identificerede en antineoplastisk forbindelse brusatol som en Nrf2-hæmmer, der øger den kemoterapeutiske effekt af cisplatin. Derudover er PI3K-hæmmere [11], [13] og Nrf2 siRNA [14] blevet brugt til at hæmme Nrf2 i kræftceller. For nylig har vi brugt en alternativ tilgang, kendt som cancer-selvmordsgenterapi, til at målrette mod kræftceller med høje Nrf2-niveauer. Nrf2-drevne lentivirale vektorer [15] indeholdende thymidinkinase (TK) overføres til cancerceller med høj ARE-aktivitet, og cellerne behandles med et pro-lægemiddel, ganciclovir (GCV). GCV metaboliseres til GCV-monophosphat, som yderligere phosphoryleres af cellulære kinaser til en giftig triphosphatform [16] (fig. 5). Dette fører til effektivt drab af ikke kun TK-holdige tumorceller, men også nabocellerne på grund af bystander-effekten [17]. ARE-reguleret TK/GCV genterapi kan forbedres yderligere ved at kombinere et cancerkemoterapeutisk middel doxorubicin til behandlingen [16], hvilket understøtter forestillingen om, at denne tilgang kunne være nyttig i forbindelse med traditionelle terapier.
Fig. 5. Selvmordsgenterapi. Konstitutiv Nrf2 nuklear akkumulering i cancerceller kan udnyttes ved at bruge Nrf2-drevet viral vektor til cancer selvmord genterapi [16]. I denne tilgang transduceres lentiviral vektor (LV), der udtrykker thymidinkinase (TK) under minimal SV40-promotor med fire ARE'er til lungeadenokarcinomceller. Høje nukleare Nrf2-niveauer fører til robust ekspression af TK gennem Nrf2-binding. Celler behandles derefter med et pro-lægemiddel, ganciclovir (GCV), som phosphoryleres af TK. Triphosphoryleret GCV forstyrrer DNA-syntesen og fører til effektivt drab af ikke kun TK-holdige tumorceller, men også nabocellerne på grund af bystander-effekten.
Nrf2 er en masterregulator, som udløser produktionen af kraftfulde antioxidanter i den menneskelige krop, som hjælper med at eliminere oxidativt stress. Forskellige antioxidantenzymer, såsom superoxiddismutase eller SOD, glutathion og katalase, aktiveres også gennem Nrf2-vejen. Desuden aktiverer visse fytokemikalier som gurkemeje, ashwagandha, bacopa, grøn te og marietidsel Nrf2. Forskningsundersøgelser har fundet det Nrf2 aktivering kan naturligt forbedre cellulær beskyttelse og genoprette balancen i den menneskelige krop.
Dr. Alex Jimenez DC, CCST Insight
Sulforaphane og dens virkninger på kræft, dødelighed, aldring, hjerne og adfærd, hjertesygdomme og mere
Isothiocyanater er nogle af de vigtigste planteforbindelser, du kan få i din kost. I denne video gør jeg det mest omfattende tilfælde til dem, der nogensinde er blevet lavet. Kort opmærksomhed span? Spring til dit yndlingsemne ved at klikke på et af nedenstående tidspunkter. Fuld tidslinje nedenfor.
Nøglesektioner:
00: 01: 14 - Kræft og dødelighed
00: 19: 04 - Aging
00: 26: 30 - Hjerne og adfærd
00: 38: 06 - Final recap
00: 40: 27 - Dosis
Fuld tidslinje:
00: 00: 34 - Introduktion af sulforaphane, et vigtigt fokus i videoen.
00: 01: 14 - Cruciferous vegetabilsk forbrug og reduktion af allårsag dødelighed.
00: 02: 12 - Prostatacancerrisiko.
00: 02: 23 - Kræftrisiko.
00: 02: 34 - Lungekræft i rygere risiko.
00: 02: 48 - Brystkræftrisiko.
00: 03: 13 - Hypotetisk: Hvad hvis du allerede har kræft? (Interventionel)
00: 03: 35 - Plausibel mekanisme, der driver kræft- og dødelighedsassosierende data.
00: 04: 38 - Sulforaphane og kræft.
00: 05: 32 - Dyrebevis, der viser stærk virkning af broccoli-spireekstrakt på udviklingen af blæretumor hos rotter.
00: 06: 06 - Virkning af direkte tilsætning af sulforaphan hos patienter med prostatacancer.
00: 07: 09 - Bioakkumulering af isothiocyanatmetabolitter i egentligt brystvæv.
00: 08: 32 - Inhibering af brystcancerstamceller.
00: 08: 53 - Historie lektion: brassicas blev etableret som at have sundhedsegenskaber selv i det gamle Rom.
00: 09: 16 - Sulforaphans evne til at øge kræftfremkaldende udskillelse (benzen, acrolein).
00: 09: 51 - NRF2 som en genetisk switch via antioxidant responselementer.
00: 10: 10 - Hvordan NRF2 aktivering øger kræftfremkaldende udskillelse via glutathion-S-konjugater.
00: 10: 34 - Spiraler øger glutathion-S-transferase og reducerer DNA-beskadigelse.
00: 11: 20 - Broccoli-spire drikker øger benzenudskillelsen med 61%.
00: 13: 31 - Broccoli spirehomogenat øger antioxidant enzymer i den øvre luftvej.
00: 15: 45 - Cruciferous vegetabilsk forbrug og dødelighed i hjertesygdomme.
00: 16: 55 - Broccoli spire pulver forbedrer blodlipider og overordnet hjertesygdomsrisiko hos type 2 diabetikere.
00: 19: 04 - Begyndelse af aldringssektionen.
00: 19: 21 - Sulforaphane beriget kost forbedrer levetiden for biller fra 15 til 30% (under visse forhold).
00: 20: 34 - Betydningen af lav inflammation for lang levetid.
00: 22: 05 - Cruciferous grøntsager og broccoli spire pulver synes at reducere et bredt udvalg af inflammatoriske markører hos mennesker.
00: 24: 14 - Musestudier tyder på, at sulforaphan kan forbedre adaptiv immunfunktion i alderdommen.
00: 25: 18 - Sulforaphane forbedrede hårvækst i en musmodel af skaldethed. Billede på 00: 26: 10.
00: 26: 30 - Begyndelse af hjerne- og adfærdssektionen.
00: 27: 18 - Effekt af broccoli-spireekstrakt på autisme.
00: 27: 48 - Virkning af glucoraphanin på skizofreni.
00: 28: 17 - Start af depression diskussion (sandsynlig mekanisme og undersøgelser).
00: 31: 21 - Mus undersøgelse ved hjælp af 10 forskellige modeller af stress-induceret depression viser sulforaphan på samme måde som fluoxetin (prozac).
00: 32: 00 - Undersøgelse viser direkte indtagelse af glucoraphanin hos mus er tilsvarende effektiv til forebyggelse af depression fra social nederlagsspændingsmodel.
00: 33: 01 - Begyndelsen af neurodegenerationsafsnittet.
00: 33: 30 - Sulforaphane og Alzheimers sygdom.
00: 33: 44 - Sulforaphane og Parkinsons sygdom.
00: 33: 51 - Sulforaphane og Hungtington's sygdom.
00: 35: 55 - Sulforaphane og neuronal plasticitet.
00: 36: 32 - Sulforaphane forbedrer læring som model af type II diabetes hos mus.
00: 37: 19 - Sulforaphane og duchenne muskeldystrofi.
00: 37: 44 - Myostatininhibering i muskel-satellitceller (in vitro).
00: 38: 06 - Late video recap: dødelighed og kræft, DNA-beskadigelse, oxidativ stress og betændelse, udslip af benzen, kardiovaskulær sygdom, type II diabetes, hjernens virkninger (depression, autisme, skizofreni, neurodegeneration), NRF2-pathway.
00: 40: 27 - Tanker om at finde ud af en dosis broccolispirer eller sulforaphane.
00: 41: 01 - Anecdoter på spiring hjemme.
00: 43: 14 - På tilberedningstemperaturer og sulforafanaktivitet.
00: 43: 45 - Gut-bakteriekonvertering af sulforaphan fra glucoraphanin.
00: 44: 24 - Tilskud fungerer bedre, når de kombineres med aktiv myrosinase fra grøntsager.
00: 44: 56 - Madlavningsteknikker og cruciferous grøntsager.
00: 46: 06 - Isothiocyanater som goitrogener.
Tak
Dette arbejde blev støttet af Akademiet i Finland, Sigrid Juselius Fonden og de finske kræftorganisationer.
Som konklusion er nuklear faktor (erythroid-afledt 2)-lignende 2, også kendt som NFE2L2 eller Nrf2, et protein, som øger produktionen af antioxidanter, som beskytter den menneskelige krop mod oxidativt stress. Som beskrevet ovenfor er stimuleringen af Nrf2-vejen ved at blive undersøgelser til behandling af sygdomme forårsaget af oxidativt stress, herunder cancer. Omfanget af vores information er begrænset til kiropraktik og rygsøjlesundhedsproblemer. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Yderligere emne diskussion: Relieving knæsmerter uden kirurgi
Knæsmerter er et velkendt symptom, der kan opstå på grund af en række knæskader og / eller tilstande, herunder sportsskader. Knæet er et af de mest komplekse led i menneskekroppen, da det er sammensat af skæret mellem fire knogler, fire ledbånd, forskellige sener, to menisci og brusk. Ifølge det amerikanske Academy of Family Physicians er de mest almindelige årsager til knæsmerter patellar subluxation, patellar tendinitis eller jumperens knæ og Osgood-Schlatter sygdom. Selv om knæsmerter sandsynligvis forekommer hos mennesker over 60 år, kan knæsmerter også forekomme hos børn og unge. Knæsmerter kan behandles hjemme efter RICE-metoderne, men alvorlige knæskader kan kræve øjeblikkelig lægehjælp, herunder kiropraktisk pleje.
DNA understøtter cirka 20,000 gener, der hver har et program til at skabe et protein eller enzym, der er nødvendigt for en sund livsstil. Hvert eneste af disse mønstre skal konstant reguleres af en slags "promotor", som styrer præcis, hvor meget af hvert stof og/eller kemikalie, der genereres, og under hvilke forhold disse også vil udvikle sig.
Ved at forbinde til en bestemt slags switch-lignende promotorområder, kendt som Antioxidant Response Element eller ARE, Nrf2 faktor�understøtter skabelseshastigheden for hundredvis af forskellige gener, som gør det muligt for cellerne at overleve under stressende omstændigheder. Disse gener genererer derefter et udvalg af antioxidantenzymer, som udvikler et forsvarsnetværk ved at neutralisere oxidanter og ved at rense de giftige biprodukter, der er efterladt i deres produktion, ud over at hjælpe med at genoprette den skade, de forårsagede.
Hvad er oxidativ stress?
Adskillige oxidanter som superoxidradikalet, eller O2-., og hydrogenperoxid eller H2O2, er blevet skabt gennem praksis med at brænde de stoffer og/eller kemikalier af, som opretholder den menneskelige krop. Den menneskelige krop besidder antioxidantenzymer, som neutraliserer og afgifter reaktive fødevarer og drikkevarer, vi indtager. Nrf2 modulerer deres produktion for at holde ligevægt og understreger efterspørgslen efter alle disse enzymer. Denne balance kan afbrydes af et par faktorer, herunder alder.
Efterhånden som vi bliver ældre, skaber den menneskelige krop mindre Nrf2, og denne delikate ligevægt kan gradvist begynde at vende sig mod den oxidative side, en tilstand der omtales som oxidativ stress. Sygdom kan også forårsage overproduktion af oxidanter. Infektioner, allergier og autoimmune lidelser kan desuden udløse vores immunceller til at skabe reaktive oxidanter, såsom O2-. , H2O2, OH og HOCl, hvor raske celler bliver beskadiget og reagerer med betændelse. Sygdomme forbundet med aldring, herunder hjerteanfald, slagtilfælde, kræft og neurodegenerative tilstande som Alzheimers sygdom, øger også udviklingen af oxidanter, hvilket genererer stress og en inflammationsreaktion.
Hvad er Nrf2 Activators?
Nrf2-proteinet, også kaldet en transkriptionsfaktor på grund af den måde, det kan understøtte og kontrollere enzymer og gener på, er det hemmelige element i en sekvens af biokemiske reaktioner i cellen, som reagerer på ændringer i kognitiv ligevægt samt oxidativ balance. Sensingselementerne i denne vej modificerer og udleder Nrf2, udløser den, så den kan spredes ind i cellekernen mod DNA'et. Nrf2 kan alternativt tænde eller slukke for de gener og enzymer, den understøtter for at beskytte cellen.
Heldigvis udvikles en række stoffer, som er Nrf2-aktivatorer, gennem indtagelse af visse planter og ekstrakter, der blev brugt for århundreder siden i traditionelle kinesiske og indianske midler. Disse fytokemikalier ser ud til at være lige så kraftige med færre bivirkninger, som de Nrf2-aktiverende farmaceutiske produkter, der bliver brugt i dag.
Nuklear faktor erythroid 2-relateret faktor, mere almindeligt kendt som Nrf2, er en transkriptionsfaktor, som beskytter cellen ved at regulere gener, enzymer og antioxidantresponser. Transkriptionsfaktorer er en type protein, der binder sig til DNA for at fremme dannelsen af specifikke stoffer og kemikalier, herunder glutathion S-transferaser eller GST'er. Nrf2-aktivering inducerer produktionen af aktive proteiner, som udviser en kraftig antioxidantkapacitet til at hjælpe med at reducere oxidativt stress.
Dr. Alex Jimenez DC, CCST Insight
Videnskaben bag Nrf2-aktivering
Da det første Nrf2-aktiverende kosttilskud blev oprettet i 2004, var minimal information kendt om funktionen af Nrf2-vejen. Der eksisterede omkring 200 aviser i litteraturen om Nrf2, også kendt som nuklear faktor-lignende 2 eller NFE2L2, og forskere var kun lige begyndt at opdage antioxidantresponsen af Nrf2 hos pattedyr. Fra 2017 er der dog blevet trykt over 9,300 akademiske forskningsstudier om denne "masterregulator".
I virkeligheden regulerer Nrf2 mange antioxidantenzymer, som ikke korrelerer med generne, i stedet tilbyder de beskyttelse mod en række stress-relaterede omstændigheder, som celler, organer og i sidste ende organismer støder på under sunde og patologiske forhold. Baseret på denne nye mængde information fra offentliggjorte akademiske forskningsstudier kan forskere nu udvikle sig bedre Nrf2 kosttilskud.
Fra 2007 har forskningsundersøgelser vist den komplekse funktion af Nrf2-vejen. Nrf2-aktivatorer har vist sig at efterligne faktorer af forskellige strukturer i den menneskelige krop. Gennem disse veje er Nrf2-aktivatorer blevet udstyret til at føle skiftende forhold i hele cellen for at holde balancen og reagere på genernes udviklende krav.
Hvorfor bruge Nrf2-aktiverende kosttilskud?
Da Nrf2-aktiveringsevner aftager med alderen i organismer, kan der begynde at forekomme ændringer. Forskningsundersøgelser har vist, at fokus på Nrf2 i celler falder med alderen, hvilket viser øgede markører for oxidativt stress. En række aldersrelaterede sygdomme som åreforkalkning og hjertekarsygdomme, gigt, kræft, fedme, type 2-diabetes, hypertension, grå stær og Alzheimers sygdom samt Parkinsons sygdomme kan udvikle sig på grund af disse ændringer. Oxidativ stress er blevet fundet med disse sundhedsproblemer.
Ved at stimulere cellens kapacitet til at øge produktionen af Nrf2-aktivatorer, Nrf2 kosttilskud kan hjælpe med at genoplive den menneskelige krops egen evne til at modvirke virkningerne af oxidativ stress. Flerumættede fedtsyrer, eller PUFA'er, er et af de lettest oxiderede molekyler, og de er særligt sårbare over for skader fra frie radikaler. Thiobarbitursyre, eller TBARS, produktion kan stige med alderen, hvilket indikerer øget oxidativ stress sammen med et fald i Nrf2-regulerede veje.
Biologisk set er geninduktion en virkelig langsom mekanisme, der generelt kræver timer at overføre gennem en vej. Som følge heraf har mange enzymer deres helt egne tænd/sluk-kontakter, som kan udløses på få minutter af forskellige regulatoriske enzymer. Forskere har udviklet proprietære sammensætninger af Nrf2-aktivatorer, som udnytter denne videnbase om aktivering. Nrf2-aktivering består ikke kun af, at Nrf2-transkriptionsfaktoren udledes fra sin inhibitor og migrerer til cellekernen, men også binding til specifikke DNA-sekvenser for at fremme cytobeskyttende genekspression, der regulerer tempoet, hvor Nrf2 tages ud af kernen.
Forståelse af elimineringsproceduren og aktiveringen af Nrf2 i den menneskelige krop har gjort det muligt for forskere at bygge kombinationer af forskellige Nrf2-aktivatorer for at opnå refleksion af gener gennem dets modulering. Kombinationen af videnbasen har sammen med den brede vifte af andre forskningsstudier bidraget til at producere Nrf2-aktivatorer til brug som kosttilskud. Omfanget af vores information er begrænset til kiropraktik og rygsøjlesundhedsproblemer. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Kurateret af Dr. Alex Jimenez
Yderligere emne diskussion: Relieving knæsmerter uden kirurgi
Knæsmerter er et velkendt symptom, der kan opstå på grund af en række knæskader og / eller tilstande, herunder sportsskader. Knæet er et af de mest komplekse led i menneskekroppen, da det er sammensat af skæret mellem fire knogler, fire ledbånd, forskellige sener, to menisci og brusk. Ifølge det amerikanske Academy of Family Physicians er de mest almindelige årsager til knæsmerter patellar subluxation, patellar tendinitis eller jumperens knæ og Osgood-Schlatter sygdom. Selv om knæsmerter sandsynligvis forekommer hos mennesker over 60 år, kan knæsmerter også forekomme hos børn og unge. Knæsmerter kan behandles hjemme efter RICE-metoderne, men alvorlige knæskader kan kræve øjeblikkelig lægehjælp, herunder kiropraktisk pleje.
Antioxidanter betegnes videnskabeligt som forbindelser, der begrænser oxidationsprocessen i menneskekroppen, at hvis de efterlades ukontrolleret, kan det skabe frie radikaler, der kan udvikle adskillige kædereaktioner, der kan forårsage cellulær skade. Heldigvis kan menneskekroppen skabe sådanne indbyggede immunmekanismer, men når montering af reaktive iltarter eller ROS ikke kan neutraliseres, forestiller man en lille flamme, der kommer ud af kontrollen, når den bliver smittet med ilt, er der risiko for skade .
For at fortsætte udvidelsen på flammens metafor er det endelige produkt, der ikke har evnen til at neutralisere virkningen af ROS eller reaktive iltarter, skade såvel som betændelse, med andre ord er menneskekroppen ret bogstaveligt i brand. Den fantastiske ting er, at der er antioxidanter, som enormt kan hjælpe med at bekæmpe dette sundhedsproblem, og denne antioxidant er glutathion. Selvom fundet i 1889, er glutathionens antioxidanteffekt blevet et af de mest interessante emner i moderne forskning.
Master of Antioxidants: Glutathione
Det kraftfulde stof er et tripeptid, der udvikles fra cystein, glutaminsyre og glycin. På grund af dets evne til at beskytte den menneskelige krop mod dannelsen af frie radikaler, kan glutathion i sidste ende hjælpe med at fremme et sundt immunsystem. Baseret på Videnskabelige rapporter i 2015, blev det bestemt, at glutathionens evne til at fungere synergistisk med peroxiredin og catalase hjælper med at beskytte celler mod hydrogenperoxid. Denne synergistiske formel fungerer mod reaktive oxygenarter, eller ROS. Glutathion, peroxidredin og catalase er væsentlige elementer i forøgelsen af cellulær homeostase, hvilket er en væsentlig proces af sunde celler, væv og organer helt og holdent.
Derudover øger glutathion den samlede immunsystemstruktur og funktion ved at udnytte den vigtige virkning på lymfocytfunktioner. Ifølge Institut for Immunokemi, korrekt supplerende niveauer af glutathion i den menneskelige krop kan i høj grad forbedre immunreaktioner. For eksempel viste to randomiserede placebokontrollerede forsøg, at den terapeutiske behandling af immunkompromitterede patienter med N-acetylcystein eller NAC resulterede i begge tilfælde i en betydelig vækst i de fleste immunologiske processer, som omfattede en hel foryngelse af naturlig mordercelleaktivitet. N-acetyl-cystein eller NAC bruger svovlet fra glutathion og kombinerer det med giftige molekyler, som derefter bliver vandopløselige og udledes i menneskekroppen.
Glutathion har også evnen til at genoplive liposyre såvel som at genbruge vitamin C og E, som er nødvendige for at initiere visse systemprocesser ved at sende elektroner til neutralisering af frie radikaler. Baseret på en undersøgelse fra PLoS ONE, glutathion ramte patienter med diabetes metillus eller T2DM og mycobacterium tuberculosis. Normalt har personer med svage immunsystemer en tendens til at vise større eksponering for M. tb eller mycobacterium tuberculosis, sygdom eller infektion. Endvidere er personer med type 2 diabetes metilllus eller T2DM to til tre gange mere tilbøjelige til TB end mennesker uden T2DM. Forskningsundersøgelsen foreslog også, at forøgelse af niveauerne af glutathion i makrofager isoleret fra patienter med T2DM førte til forbedret kontrol af M.Tb sygdom eller infektion. Disse resultater viser, at lavere niveauer af glutathion hos patienter med T2DM bidrager til en øget chance for M. tb sygdom eller infektion. Desuden afhængig af Dietro Ghezzi i Brighton og Sussex Medical School, oxidativ stress kan i sidste ende forårsage dårlig immunsystem struktur og funktion.
Heldigvis spiller glutathion en afgørende rolle i styrkelse og kontrol af immunitet. Som eksempel er glutathion essentiel for medfødte og adaptive processer i immunsystemet, herunder T-lymfocytproliferation, fagocytisk aktivitet af polymorfonukleære neutrofile og dendritiske cellefunktioner, hvilket kan være fundamental, fordi disse er sammensat af antigenpræsenterende celler . Cellmediteret immunitet indbefatter proteinantigener, som oprindeligt begynder at degenerere i de endocytiske vesikler af makrofager og dendritiske celler, derfor er de mindre peptider på overfladen demonstreret for at aktivere proliferation af antigenspecifikke T-celler. Derudover hjælper glutathion med dannelsen af cytokiner, og det er nødvendigt at opretholde interferon-gamma-produktion ved hjælp af dendritiske celler, hvilket er vigtigt for at beskytte mod intracellulære patogener, herunder mykobakterier.
N-acetyl-cystein eller NAC, der videnskabeligt er omtalt som forløber for glutathion, er også en meget kraftig cellulær antioxidant, der anvendes som antioxidant med fri radikal. Fælles for sin rolle i afværge acetaminophen toksicitet, NAC, eller �N-acetyl-cystein, har vist sig at have adskillige sundheds- og wellnessfordele. Ifølge Cell Journal, NAC hjælper med at støtte en sund inflammatorisk reaktion og kan positivt påvirke menneskelige sigt og tidlige arbejde. Forskningsundersøgelsen konkluderede, at hos kvinder med tidligere tidligere fødsel og bakteriel vaginose, blev 0.6 gram NAC pr. Dag taget oralt sammen med progesteron efter uge 16 af graviditeten afskærmet mod fornyet fødsel og forbedret neonatale udfald. Afslutningsvis blev NACs positive virkninger på muskelopbygning også påvist. Efter tre minutters vedvarende sammentrækninger var der en forbedret 15-procent, hvilket viste, hvordan NAC spiller en grundlæggende rolle i forbedringen af muskelopbygningen og reducerer den samlede træthed under arbejdet.
Forskere opdagede også, at NAC, eller N-acetyl-cystein, kan gavne dem, der har polycystisk ovariesyndrom eller PCOS. PCOS, eller polycystisk ovariesyndrom, er en almindelig endokrin kirtelrelateret sygdom, som rammer cirka 5 til 10 procent af kvinder i den reproduktive alder. Hos sådanne patienter er der større risiko for at opleve metabolisk syndrom, hvor brugen af NAC hjalp med at genoprette sunde insulinniveauer og følsomhed.
Dr. Alex Jimenez's Insight
Glutathion er blevet omtalt som "master of antioxidants" på grund af dets grundlæggende rolle i at opnå og opretholde helbred og wellness. Mens menneskekroppen er i stand til at producere sin egen glutathion, kan dårlig ernæring, forurening, toksiner, overdreven brug af stoffer og / eller medicin, stress, traume, aldring, sygdom og stråling alle reducere vores naturlige niveauer af glutathion. Dette kan igen gøre individer mere modtagelige for celleskader fra oxidativ stress, frie radikaler, infektioner og kræft. Glutathiontilskud kan derfor have enorme fordele på den menneskelige krop. Sammen med alternative behandlingsmuligheder, såsom kiropraktisk pleje, kan glutathioniveauer igen reguleres for at forbedre trivsel.
Derudover har sundhedspersonale foreslået at implementere brugen af glutathiontilskud sammen med andre alternative behandlingsmuligheder, som f.eks kiropraktik pleje, for yderligere at forbedre den generelle sundhed og velvære. Antioxidanter er vigtige for at opretholde maksimal velvære såvel som at hæmme kædereaktionen af frie radikaler, der forårsager cellers skade eller skade. Kraftige antioxidanter som glutathion, som tidligere nævnt, hjælper i sidste ende med at regulere udviklingen af disse frie radikaler og give et sundere immunsystem respons. Forskningsundersøgelser har fundet det kiropraktik pleje kan også spille en afgørende rolle i denne proces, hvilket naturligt øger antioxidants aktivitet i den menneskelige krop. Kiropraktisk pleje er en sikker og effektiv behandlingsmetode, der anvender spinaljusteringer og manuelle manipulationer til at korrigere spinal forskydninger eller subluxationer for at gøre det muligt for den menneskelige krop naturligt at helbrede sig uden brug af stoffer / medicin og / eller kirurgiske indgreb.
Endelig demonstrerer antioxidanter deres biologiske egenskaber gennem en lang række sundhedsmæssige fordele, som måske er nødvendige nu mere end nogensinde med det stadigt stigende angreb af stress, sygdom og forurening i vores moderne verden, som alle bidrager til celleskade og/eller beskadigelse . Glutathion og dets forløber, NAC, eller N-acetyl-cystein, fortsætter med at vise deres stærke status inden for antioxidanter. Sammen med alternative behandlingsmuligheder, såsom kiropraktisk behandling, kan folk drage fordel af alle de fordele, som denne kraftfulde antioxidant har at tilbyde. Omfanget af vores information er begrænset til kiropraktik samt til rygmarvsskader og -tilstande. For at diskutere emnet, er du velkommen til at spørge Dr. Jimenez eller kontakte os på�915-850-0900 .
Kurateret af Dr. Alex Jimenez
Yderligere emner: Rygsmerter
Rygsmerte er en af de mest udbredte årsager til handicap og savnede dage på arbejdspladsen over hele verden. Faktisk er rygsmerter blevet tilskrevet som den næst mest almindelige årsag til doktorkontorbesøg, der kun overstiger luftvejsinfektioner. Ca. 80 procent af befolkningen vil opleve en form for rygsmerter mindst én gang i hele deres liv. Ryggraden er en kompleks struktur bestående af knogler, led, ledbånd og muskler, blandt andet blødt væv. På grund af dette skader og / eller forværrede forhold, som f.eks herniated diske, kan i sidste ende føre til symptomer på rygsmerter. Sportsskader eller personskader er ofte den hyppigste årsag til rygsmerter, men nogle gange kan de enkleste bevægelser have smertefulde resultater. Heldigvis kan alternative behandlingsmuligheder, såsom kiropraktisk pleje, hjælpe lindring af rygsmerter ved brug af rygtilpasninger og manuelle manipulationer, der i sidste ende forbedrer smertelindring.
Videnskabsbaseret kiropraktor Dr. Alexander Jimenez kigger på oxidativt stress, hvad det er, hvordan det påvirker kroppen og antioxidantforsvaret for at afhjælpe situationen.
Esra Birben PhD,1 Umit Murat Sahiner MD,1 Cansin Sackesen MD,1 Serpil Erzurum MD,2 og Omer Kalayci, MD1
Abstrakt: Reaktive oxygenarter (ROS) produceres af levende organismer som et resultat af normal cellulær metabolisme og miljøfaktorer, såsom luftforurenende stoffer eller cigaretrøg. ROS er meget reaktive molekyler og kan beskadige cellestrukturer såsom kulhydrater, nukleinsyrer, lipider og proteiner og ændre deres funktioner. Skiftet i balancen mellem oxidanter og antioxidanter til fordel for oxidanter kaldes �oxidativt stress.� Regulering af reducerende og oxiderende (redox) tilstand er afgørende for cellernes levedygtighed, aktivering, proliferation og organfunktion. Aerobe organismer har integrerede antioxidantsystemer, som omfatter enzymatiske og ikke-enzymatiske antioxidanter, der normalt er effektive til at blokere skadelige virkninger af ROS. Men under patologiske tilstande kan antioxidantsystemerne blive overvældet. Oxidativt stress bidrager til mange patologiske tilstande og sygdomme, herunder cancer, neurologiske lidelser, åreforkalkning, hypertension, iskæmi/perfusion, diabetes, akut respiratorisk distress-syndrom, idiopatisk lungefibrose, kronisk obstruktiv lungesygdom og astma. I denne gennemgang opsummerer vi de cellulære oxidant- og antioxidantsystemer og diskuterer de cellulære virkninger og mekanismer af oxidativ stress.
Reaktive oxygenarter (ROS) produceres af levende organismer som et resultat af normal cellulær metabolisme. Ved lave til moderate koncentrationer fungerer de i fysiologiske celleprocesser, men ved høje koncentrationer producerer de negative modifikationer af cellekomponenter, såsom lipider, proteiner og DNA.1�6 Skiftet i balance mellem oxidant/antioxidant til fordel for oxidanter. kaldes �oxidativt stress.� Oxidativt stress bidrager til mange patologiske tilstande, herunder kræft, neurologiske lidelser,7�10 åreforkalkning, hypertension, iskæmi/perfusion,11�14 diabetes, akut respiratorisk distress-syndrom, idiopatisk lungefibrose, kronisk obstruktiv sygdom ,15 og astma.16�21 Aerobe organismer har integrerede antioxidantsystemer� som omfatter enzymatiske og ikke-enzymatiske antioxidanter, der normalt er effektive til at blokere skadelige virkninger af ROS. Men under patologiske tilstande kan antioxidantsystemerne blive overvældet. I denne gennemgang opsummerer vi de cellulære oxidant- og antioxidantsystemer og regulering af den reducerende og oxiderende (redox) tilstand i sundheds- og sygdomstilstande.
OXIDANTER
Endogene kilder til ROS
ROS produceres ud fra molekylær oxygen som et resultat af normal cellulær metabolisme. ROS kan opdeles i 2 grupper: frie radikaler og ikke-radikaler. Molekyler, der indeholder en eller flere uparrede elektroner og dermed giver reaktivitet til molekylet, kaldes frie radikaler. Når 2 frie radikaler deler deres uparrede elektroner, dannes der ikke-radikale former. De 3 store ROS, der er af fysiologisk betydning, er superoxidanion (O22.), hydroxylradikal (OH) og hydrogenperoxid (H2O2). ROS er opsummeret i tabel 1.
Superoxidanion dannes ved tilsætning af 1 elektron til det molekylære oxygen.22 Denne proces medieres af nikotin-adenindinukleotidphosphat [NAD(P)H]-oxidase eller xanthinoxidase eller af mitokondrielt elektrontransportsystem. Det vigtigste sted for fremstilling af superoxidanion er mitokondrierne, cellens maskineri til at producere adenosintrifosfat. Normalt overføres elektroner gennem mitokondriel elektrontransportkæde til reduktion af ilt til vand, men cirka 1 til 3% af alle elektroner lækker fra systemet og producerer superoxid. NAD(P)H-oxidase findes i polymorfonukleære leukocytter, monocytter og makrofager. Ved fagocytose producerer disse celler et udbrud af superoxid, der fører til bakteriedræbende aktivitet. Superoxid omdannes til hydrogenperoxid ved påvirkning af superoxiddismutaser (SODs, EC 1.15.1.1). Hydrogenperoxid diffunderer let hen over plasmamembranen. Hydrogenperoxid produceres også af xanthinoxidase, aminosyreoxidase og NAD(P)H-oxidase�23,24 og i peroxisomer ved forbrug af molekylært oxygen i metaboliske reaktioner. I en række reaktioner kaldet Haber�Weiss- og Fenton-reaktioner kan H2O2 nedbrydes til OH2 i nærværelse af transmissionsmetaller som Fe21 eller Cu21.25
Fe31 +�.O2�?Fe2 +�O2 Haber Weiss
Fe2 +�H2O2�?Fe3 +�OH�+ .OH Fenton-reaktion
O 2 i sig selv kan også reagere med H2 O2 og generere OH�.26,27 Hydroxyl-radikal er den mest reaktive af ROS og kan beskadige proteiner, lipider og kulhydrater og DNA. Det kan også starte lipidperoxidation ved at tage en elektron fra flerumættede fedtsyrer.
Granulocytiske enzymer udvider yderligere reaktiviteten af H2O2 via eosinofil peroxidase og myeloperoxidase (MPO). I aktiverede neutrofiler forbruges H2O2 af MPO. I nærvær af chloridion omdannes H2O2 til hypochlorsyrling (HOCl). HOCl er meget oxidativt og spiller en vigtig rolle i at dræbe patogenerne i luftvejene.28 Imidlertid kan HOCl også reagere med DNA og inducere DNA-protein-interaktioner og producere pyrimidinoxidationsprodukter og tilføje chlorid til DNA-baser.29,30 Eosinophil peroxidase og MPO bidrager også til det oxidative stress ved at modificere proteiner ved halogeneringer, nitrering og proteintværbindinger via tyrosylradikaler.31�33
Andre oxygen-afledte frie radikaler er peroxyl-radikaler (ROO$ ). Den enkleste form for disse radikaler er hydroperoxylradikal (HOO$) og spiller en rolle i fedtsyreperoxidation. Frie radikaler kan udløse lipidperoxidationskædereaktioner ved at abstrahere et brintatom fra et sidekæde-methylencarbon. Lipid radikalet reagerer derefter med oxygen for at producere peroxyl radikal. Peroxyl-radikal starter en kædereaktion og omdanner flerumættede fedtsyrer til lipidhydroperoxider. Lipidhydroperoxider er meget ustabile og nedbrydes let til sekundære produkter, såsom aldehyder (såsom 4-hydroxy-2,3-nonenal) og malondialdehyder (MDA). Isoprostaner er en anden gruppe af lipidperoxidationsprodukter, der genereres via peroxidation af arachidonsyre, og som også har vist sig at være forhøjet i plasma og åndedrætskondensater fra astmatikere.34,35 Peroxidation af lipider forstyrrer cellemembranernes integritet og fører til omarrangering af membran struktur.
Hydrogenperoxid, superoxidradikal, oxideret glutathion (GSSG), MDA'er, isoprostaner, carbonyler og nitrotyrosin kan let måles fra plasma-, blod- eller bronkoalveolære skylleprøver som biomarkører for oxidation ved standardiserede analyser.
Eksogen kilde til oxidanter
Cigaretrøg
Cigaretrøg indeholder mange oxidanter og frie radikaler og organiske forbindelser, såsom superoxid og nitrogenoxid.36 Derudover aktiverer indånding af cigaretrøg i lungen også nogle endogene mekanismer, såsom ophobning af neutrofiler og makrofager, som yderligere øger oxidantskaden .
Ozoneksponering
Ozoneksponering kan forårsage lipidperoxidation og inducere tilstrømning af neutrofiler i luftvejsepitelet. Kortvarig eksponering for ozon forårsager også frigivelse af inflammatoriske mediatorer, såsom MPO, eosinofile kationiske proteiner og også lactatdehydrogenase og albumin.37 Selv hos raske forsøgspersoner forårsager ozoneksponering en reduktion i lungefunktioner.38 Cho et al39 har vist, at partikler (blanding af faste partikler og flydende dråber suspenderet i luften) katalyserer reduktionen af ilt.
hyperoxi
Hyperoksi refererer til tilstande med højere iltniveauer end normalt partialtryk af ilt i lungerne eller andet kropsvæv. Det fører til større produktion af reaktive oxygen- og nitrogenarter.40,41
Ioniserende stråling
Ioniserende stråling, i nærvær af O2, omdanner hydroxylradikal, superoxid og organiske radikaler til hydrogenperoxid og organiske hydroperoxider. Disse hydroperoxidarter reagerer med redoxaktive metalioner, såsom Fe og Cu, via Fenton-reaktioner og inducerer dermed oxidativ stress.42,43 Narayanan et al44 viste, at fibroblaster, der blev udsat for alfapartikler, havde signifikante stigninger i intracellulær O2 2. og H2O2 produktion via plasmamembranbundet NADPH-oxidase.44 Signaltransduktionsmolekyler, såsom ekstracellulær signalreguleret kinase 1 og 2 (ERK1/2), c-Jun N-terminal kinase (JNK) og p38, og transkriptionsfaktorer, som f.eks. aktivatorprotein-1 (AP-1), nuklear faktor-kB (NF-kB) og p53 aktiveres, hvilket resulterer i ekspression af strålingsrespons-relaterede gener.45-50 Ultraviolet A (UVA) fotoner udløser oxidative reaktioner ved excitation af endogene fotosensibilisatorer, såsom porphyriner, NADPH-oxidase og riboflaviner. 8-Oxo-7,8-dihydroguanin (8-oxoGua) er det vigtigste UVA-medierede DNA-oxidationsprodukt, der dannes ved oxidation af OH-radikal, 1-elektronoxidanter og singlet oxygen, der hovedsageligt reagerer med guanin.51 Dannelsen af guanin radikal kation i isoleret DNA har vist sig effektivt at opstå gennem den direkte effekt af ioniserende stråling.52,53 Efter udsættelse for ioniserende stråling falder det intracellulære niveau af glutathion (GSH) i kort tid, men stiger derefter igen.54
Heavy Metal ioner
Tungmetalioner, såsom jern, kobber, cadmium, kviksølv, nikkel, bly og arsen, kan inducere dannelse af reaktive radikaler og forårsage cellulær skade via udtømning af enzymaktiviteter gennem lipidperoxidation og reaktion med nukleare proteiner og DNA.55
En af de vigtigste mekanismer for metal-medieret frie radikaler er via en Fenton-type reaktion. Superoxidion og hydrogenperoxid kan interagere med overgangsmetaller, såsom jern og kobber, via den metalkatalyserede Haber�Weiss/Fenton-reaktion for at danne OH-radikaler.
Udover mekanismerne af Fenton-typen og Haber�Weiss-typen kan visse metalioner reagere direkte med cellulære molekyler for at generere frie radikaler, såsom thiolradikaler, eller inducere cellesignalveje. Disse radikaler kan også reagere med andre thiolmolekyler for at danne O22.. O22. omdannes til H2O2, hvilket forårsager yderligere iltradikalerdannelse. Nogle metaller, såsom arsenit, inducerer ROS-dannelse indirekte ved aktivering af radikalproducerende systemer i celler.56
Arsen er et meget giftigt grundstof, der producerer en række ROS, herunder superoxid (O2 2), singlet oxygen (1O2), peroxyl radikal (ROO ), nitrogenoxid (NO ), hydrogenperoxid (H2O2) og dimethylarsiniske peroxyl radikaler [( CH3)2AsOO ].57�59 Arsen (III)-forbindelser kan hæmme antioxidantenzymer, især de GSH-afhængige enzymer, såsom glutathion-S-transferaser (GST'er), glutathionperoxidase (GSH-Px) og GSH-reduktase, via binding - ing til deres sulfhydryl (�SH) grupper.60,61
Bly øger lipidperoxidation.62 Betydelige fald i aktiviteten af vævs-SOD og hjerne-GPx er blevet rapporteret efter blyeksponering.63,64 Erstatning af zink, som tjener som en co-faktor for mange enzymer med bly, fører til inaktivering af sådanne enzymer. Blyeksponering kan forårsage hæmning af GST ved at påvirke thioler i væv.
ROS genereret af metalkatalyserede reaktioner kan modificere DNA-baser. Tre basesubstitutioner, G/C, G/T og C/T, kan forekomme som følge af oxidativ beskadigelse af metalioner, såsom Fe21, Cu21 og Ni21. Reid et al65 viste, at G/C overvejende blev produceret af Fe21, mens C/T-substitution var af Cu21 og Ni21.
ANTIOXIDANTER
Den menneskelige krop er udstyret med en række antioxidanter, der tjener til at opveje virkningen af oxidanter. For alle praktiske formål kan disse opdeles i 2 kategorier: enzymatiske (tabel 2) og ikke-enzymatiske (tabel 3).
Enzymatiske antioxidanter
De vigtigste enzymatiske antioxidanter i lungerne er SOD'er (EC 1.15.1.11), katalase (EC 1.11.1.6) og GSH-Px (EC 1.11.1.9). Ud over disse vigtige enzymer, andre antioxidanter, herunder hæm-oxygenase-1 (EC 1.14.99.3) og redoxproteiner, såsom thioredoxiner (TRX'er, EC 1.8.4.10), peroxiredoxiner (PRX'er, EC 1.11.1.15) og glutaredoxiner , har også vist sig at spille en afgørende rolle i det pulmonale antioxidantforsvar.
Da superoxid er det primære ROS produceret fra en række forskellige kilder, er dets dismutation af SOD af primær betydning for hver celle. Alle 3 former for SOD, det vil sige CuZn-SOD, Mn-SOD og EC-SOD, er bredt udtrykt i den menneskelige lunge. Mn-SOD er lokaliseret i mitokondriermatrixen. EC-SOD er primært lokaliseret i den ekstracellulære matrix, især i områder, der indeholder store mængder af type I kollagenfibre og omkring pulmonale og systemiske kar. Det er også blevet påvist i bronkialepitel, alveolært epitel og alveolære makrofager.66,67 Samlet set menes CuZn-SOD og Mn-SOD generelt at fungere som bulkfangere af superoxidradikaler. Det relativt høje EC-SOD-niveau i lungen med dens specifikke binding til de ekstracellulære matrixkomponenter kan repræsentere en fundamental komponent i lungematrixbeskyttelse.68
H2O2, der produceres ved virkningen af SOD'er eller virkningen af oxidaser, såsom xanthinoxidase, reduceres til vand af katalase og GSH-Px. Catalase eksisterer som en tetramer sammensat af 4 identiske monomerer, som hver indeholder en hæmgruppe på det aktive sted. Nedbrydning af H2O2 opnås via omdannelsen mellem 2 konformationer af katalase-ferricatalase (jern koordineret til vand) og forbindelse I (jern kompleksbundet med et oxygenatom). Catalase binder også NADPH som en reducerende ækvivalent for at forhindre oxidativ inaktivering af enzymet (dannelse af forbindelse II) af H2O2, når det reduceres til vand.69
Enzymer i redoxcyklussen, der er ansvarlige for reduktionen af H2O2 og lipidhydroperoxider (genereret som et resultat af membranlipidperoxidation), omfatter GSH-Pxs.70 GSH-Pxs er en familie af tetramere enzymer, der indeholder den unikke aminosyre selenocystein i aktive steder og anvende lavmolekylære thioler, såsom GSH, til at reducere H2O2 og lipidperoxider til deres tilsvarende alkoholer. Fire GSH-Px'er er blevet beskrevet, kodet af forskellige gener: GSH-Px-1 (cellulær GSH-Px) er allestedsnærværende og reducerer H2O2 og fedtsyreperoxider, men ikke esterificerede peroxyllipider.71 Esterificerede lipider reduceres af membranbundet GSH -Px-4 (phospholipidhydroperoxid GSH-Px), som kan bruge flere forskellige lavmolekylære thioler som reducerende ækvivalenter. GSH-Px-2 (gastrointestinal GSH-Px) er lokaliseret i gastrointestinale epitelceller, hvor det tjener til at reducere diætperoxider.72 GSH-Px-3 (ekstracellulær GSH-Px) er det eneste medlem af GSH-Px-familien, der er bosat i det ekstracellulære rum og menes at være et af de vigtigste ekstracellulære antioxidantenzym hos pattedyr. Af disse er ekstracellulær GSH-Px mest udbredt undersøgt i den menneskelige lunge.73
Derudover er bortskaffelse af H2O2 tæt forbundet med flere thiol-holdige enzymer, nemlig TRX'er (TRX1 og TRX2), thioredoxinreduktaser (EC 1.8.1.9) (TRR'er), PRX'er (som er thioredoxinperoxidaser) og glutaredoxiner.74
To TRX'er og TRR'er er blevet karakteriseret i humane celler, der eksisterer i både cytosol og mitokondrier. I lungen udtrykkes TRX og TRR i bronchial og alveolært epitel og makrofager. Seks forskellige PRX'er er blevet fundet i humane celler, der adskiller sig i deres ultrastrukturelle kompartmentalisering. Eksperimentelle undersøgelser har afsløret vigtigheden af PRX VI i beskyttelsen af alveolært epitel. Human lunge udtrykker alle PRX'er i bronchial epitel, alveole epitel og makrofager.75 PRX V har for nylig vist sig at fungere som en peroxynitrit reduktase,76 hvilket betyder, at det kan fungere som en potentiel beskyttende forbindelse i udviklingen af ROS-medieret lungeskade .77
Fælles for disse antioxidanter er kravet om NADPH som reducerende ækvivalent. NADPH opretholder katalase i den aktive form og bruges som en cofaktor af TRX og GSH-reduktase (EC 1.6.4.2), som omdanner GSSG til GSH, et co-substrat for GSH-Pxs. Intracellulær NADPH genereres igen ved reduktion af NADP1 af glucose-6-phosphat-dehydrogenase, det første og hastighedsbegrænsende enzym i pentose-phosphat-vejen, under omdannelsen af glucose-6-phosphat til 6-phosphogluconolacton. Ved at generere NADPH er glucose-6-phosphat-dehydrogenase en kritisk determinant for cytosolisk GSH-bufferkapacitet (GSH/GSSG) og kan derfor betragtes som et væsentligt, regulerende antioxidantenzym.78,79
GST'er (EC 2.5.1.18), en anden antioxidantenzymfamilie, inaktiverer sekundære metabolitter, såsom umættede aldehyder, epoxider og hydroperoxider. Tre hovedfamilier af GST'er er blevet beskrevet: cytosolisk GST, mitokondriel GST,80,81 og membranassocieret mikrosomal GST, der har en rolle i eicosanoid- og GSH-metabolisme.82 Syv klasser af cytosolisk GST er identificeret i pattedyr, betegnet Alpha, Mu, Pi, Sigma, Theta, Omega og Zeta.83�86 Under ikke-stressede tilstande interagerer klasse Mu og Pi GST'er med henholdsvis kinaserne Ask1 og JNK og hæmmer disse kinaser.87�89 Det er vist, at GSTP1 dissocierer fra JNK som reaktion på oxidativt stress.89 GSTP1 interagerer også fysisk med PRX VI og fører til genvinding af PRX-enzymaktivitet via glutathionylering af det oxiderede protein.90
Ikke-enzymatiske antioxidanter
Ikke-enzymatiske antioxidanter omfatter forbindelser med lav molekylvægt, såsom vitaminer (vitamin C og E), b-caroten, urinsyre og GSH, et tripeptid (Lg-glutamyl-L-cysteinyl-L-glycin), der omfatter en thiol ( sulfhydryl) gruppe.
Vitamin C (ascorbinsyre)
Vandopløseligt C-vitamin (ascorbinsyre) giver intracellulær og ekstracellulær vandfase antioxidantkapacitet primært ved at opfange frie iltradikaler. Det konverterer vitamin E frie radikaler tilbage til vitamin E. Dets plasmaniveauer har vist sig at falde med alderen.91,92
Vitamin E (a-Tocopherol)
Lipidopløseligt vitamin E er koncentreret i det hydrofobe indre sted af cellemembranen og er det vigtigste forsvar mod oxidant-induceret membranskade. E-vitamin donerer elektron til peroxyl-radikal, som produceres under lipidperoxidation. a-Tocopherol er den mest aktive form for vitamin E og den vigtigste membranbundne antioxidant i cellen. E-vitamin udløser apoptose af kræftceller og hæmmer frie radikaler.93
Glutathion
GSH er meget rigeligt i alle celle-rum og er den vigtigste opløselige antioxidant. GSH/GSSG-forholdet er en væsentlig determinant for oxidativ stress. GSH viser sine antioxidante virkninger på flere måder.94 Det afgifter hydrogenperoxid og lipidperoxider via virkning af GSH-Px. GSH donerer sin elektron til H2O2 for at reducere den til H2O og O2. GSSG reduceres igen til GSH af GSH-reduktase, der bruger NAD(P)H som elektrondonor. GSH-Px'er er også vigtige for beskyttelsen af cellemembranen mod lipidperoxidation. Reduceret glutathion donerer protoner til membranlipider og beskytter dem mod oxidantangreb.95
GSH er en cofaktor for flere afgiftende enzymer, såsom GSH-Px og transferase. Det har en rolle i at omdanne C- og E-vitamin tilbage til deres aktive former. GSH beskytter celler mod apoptose ved at interagere med proapoptotiske og antiapoptotiske signalveje.94 Det regulerer og aktiverer også flere transkriptionsfaktorer, såsom AP-1, NF-kB og Sp-1.
Carotenoider (b-caroten)
Carotenoider er pigmenter, der findes i planter. Det har primært vist sig, at b-caroten reagerer med peroxyl (ROO ), hydroxyl (OH) og superoxid (O22.) radikaler.96 Carotenoider viser deres antioxidantvirkninger ved lavt iltpartialtryk, men kan have prooxidantvirkninger ved højere oxygen koncentrationer.97 Både carotenoider og retinoinsyrer (RA'er) er i stand til at regulere transkriptionsfaktorer.98 b-Caroten hæmmer den oxidant-inducerede NF-kB-aktivering og interleukin (IL)-6 og tumornekrosefaktor-a-produktion. Carotenoider påvirker også apoptose af celler. Antiproliferative virkninger af RA er blevet vist i flere undersøgelser. Denne effekt af RA medieres hovedsageligt af retinsyrereceptorer og varierer mellem celletyper. I brystkarcinomceller blev retinsyrereceptor vist at udløse væksthæmning ved at inducere cellecyklusstop, apoptose eller begge dele.99,100
EFFEKTEN AF OXIDATIV STRESS: GENETISKE, FYSIOLOGISKE OG BIOKEMISKE MEKANISMER
Oxidativt stress opstår, når balancen mellem antioxidanter og ROS forstyrres på grund af enten udtømning af antioxidanter eller ophobning af ROS. Når oxidativt stress opstår, forsøger cellerne at modvirke oxidanteffekterne og genoprette redoxbalancen ved aktivering eller dæmpning af gener, der koder for defensive enzymer, transkriptionsfaktorer og strukturelle proteiner.101,102 Forholdet mellem oxideret og reduceret glutathion (2GSH/GSSG) er ét. af de vigtige determinanter for oxidativt stress i kroppen. Højere produktion af ROS i kroppen kan ændre DNA-strukturen, resultere i modifikation af proteiner og lipider, aktivering af flere stress-inducerede transkriptionsfaktorer og produktion af pro-inflammatoriske og anti-inflammatoriske cytokiner.
Virkninger af oxidativ stress på DNA
ROS kan føre til DNA-modifikationer på flere måder, hvilket involverer nedbrydning af baser, enkelt- eller dobbeltstrengede DNA-brud, purin, pyrimidin eller sukkerbundne modifikationer, mutationer, deletioner eller translokationer og tværbinding med proteiner. De fleste af disse DNA-modifikationer (fig. 1) er yderst relevante for carcinogenese, aldring og neurodegenerative, kardiovaskulære og autoimmune sygdomme. Tobaksrøg, redoxmetaller og nonredox-metaller, såsom jern, cadmium, krom og arsen, er også involveret i carcinogenese og aldring ved at generere frie radikaler eller binde sig til thiolgrupper. Dannelse af 8-OH-G er den bedst kendte DNA-skade, der opstår via oxidativ stress og er en potentiel biomarkør for carcinogenese.
Promotorregioner af gener indeholder konsensussekvenser for transkriptionsfaktorer. Disse transkriptionsfaktorbindingssteder indeholder GC-rige sekvenser, der er modtagelige for oxidantangreb. Dannelse af 8-OH-G DNA i transkriptionsfaktorbindingssteder kan modificere binding af transkriptionsfaktorer og dermed ændre ekspressionen af beslægtede gener, som det er blevet vist for AP-1 og Sp-1 målsekvenser.103 Udover 8-OH-G, 8,59-cyclo-29-deoxyadenosin (cyclo-dA) har også vist sig at hæmme transkription fra et reportergen i et cellesystem, hvis det er placeret i en TATA-boks.104 Det TATA-bindende protein initierer transkription ved at ændre bøjningen af DNA . Bindingen af TATA-bindende protein kan være svækket af tilstedeværelsen af cyclo-dA.
Oxidativ stress forårsager ustabilitet i mikrosatellitregioner (korte tandemgentagelser). Redox-aktive metalioner, hydroxylradikaler øger mikrosatellit-ustabiliteten.105 Selvom enkeltstrengede DNA-brud forårsaget af oxidantskade let kan tolereres af celler, kan dobbeltstrengede DNA-brud fremkaldt af ioniserende stråling være en væsentlig trussel for cellens overlevelse.106
Methylering ved CpG-øer i DNA er en vigtig epigenetisk mekanisme, der kan resultere i gendæmpning. Oxidation af 5-MeCyt til 5-hydroxymethyl uracil (5-OHMeUra) kan ske via deaminerings-/oxidationsreaktioner af thymin eller 5-hydroxymethylcytosin-mellemprodukter.107 Ud over den modulerende genekspression ser DNA-methylering også ud til at påvirke kromatinorganisationen.108 Afvigende DNA-methyleringsmønstre induceret af oxidative angreb påvirker også DNA-reparationsaktivitet.
Virkninger af oxidativ stress på lipider
ROS kan inducere lipidperoxidation og forstyrre membranens lipid-dobbeltlagsarrangement, der kan inaktivere membranbundne receptorer og enzymer og øge vævspermeabiliteten.109 Produkter af lipidperoxidation, såsom MDA og umættede aldehyder, er i stand til at inaktivere mange cellulære proteiner ved at danne proteinkryds. -bindinger.110�112 4-Hydroxy-2-nonenal forårsager udtømning af intracellulært GSH og inducerer peroxidproduktion,113,114 aktiverer epidermal vækstfaktorreceptor115 og inducerer fibronectinproduktion.116 Lipidperoxidationsprodukter, såsom isoprostaner og thiobarbiturinsyre-reaktive stoffer , er blevet brugt som indirekte biomarkører for oxidativt stress, og øgede niveauer blev vist i udåndingskondensat eller bronkoalveolær skyllevæske eller lunge hos patienter med kronisk obstruktiv lungesygdom eller rygere.117�119
Effekter af oxidativ stress på proteiner
ROS kan forårsage fragmentering af peptidkæden, ændring af elektrisk ladning af proteiner, tværbinding af proteiner og oxidation af specifikke aminosyrer og derfor føre til øget modtagelighed for proteolyse ved nedbrydning af specifikke proteaser.120 Cystein- og methioninrester i proteiner er særligt mere modtagelige for oxidation.121 Oxidation af sulfhydrylgrupper eller methioninrester af proteiner forårsager konformationelle ændringer, proteinudfoldning og nedbrydning.8,121�123 Enzymer, der har metaller på eller tæt på deres aktive steder, er især mere følsomme over for metalkatalyseret oxidation. Oxidativ modifikation af enzymer har vist sig at hæmme deres aktiviteter.124,125
I nogle tilfælde kan specifik oxidation af proteiner finde sted. For eksempel kan methionin være oxideret methioninsulfoxid126 og phenylalanin til o-tyrosin127; sulfhydrylgrupper kan oxideres til dannelse af disulfidbindinger;128 og carbonylgrupper kan indføres i proteiners sidekæder. Gammastråler, metalkatalyseret oxidation, HOCl og ozon kan forårsage dannelse af carbonylgrupper.129
Effekter af oxidativ stress på signaltransduktion
ROS kan inducere ekspression af flere gener involveret i signaltransduktion.1,130 Et højt forhold for GSH/GSSG er vigtigt for beskyttelsen af cellen mod oxidativ skade. Afbrydelse af dette forhold forårsager aktivering af redoxfølsomme transkriptionsfaktorer, såsom NF-kB, AP-1, nuklear faktor af aktiverede T-celler og hypoxi-inducerbar faktor 1, der er involveret i det inflammatoriske respons. Aktivering af transkriptionsfaktorer via ROS opnås ved signaltransduktionskaskader, der transmitterer informationen udefra til indersiden af cellen. Tyrosinkinasereceptorer, de fleste af vækstfaktorreceptorerne, såsom epidermal vækstfaktorreceptor, vaskulær endotelvækstfaktorreceptor og receptor for blodpladeafledt vækstfaktor, proteintyrosinphosphataser og serin/threoninkinaser er mål for ROS.131�133 Ekstracellulære signalregulerede kinaser, JNK og p38, som er medlemmer af mitogenaktiveret proteinkinasefamilie og involveret i adskillige processer i cellen, herunder proliferation, differentiering og apoptose, kan også reguleres af oxidanter.
Under oxidative stressforhold gennemgår cysteinrester i DNA-bindingsstedet i c-Jun, nogle AP-1-underenheder og inhiberende kB-kinase reversibel S-glutathiolering. Glutaredoxin og TRX er blevet rapporteret at spille en vigtig rolle i reguleringen af redox-følsomme signalveje, såsom NF-kB og AP-1, p38 mitogen-aktiveret proteinkinase og JNK.134�137
NF-kB kan aktiveres som reaktion på oxidative stresstilstande, såsom ROS, frie radikaler og UV-bestråling.138 Fosforylering af IkB frigør NF-kB og tillader det at komme ind i kernen for at aktivere gentranskription.139 En række kinaser har blevet rapporteret at phosphorylere IkB'er ved serinresterne. Disse kinaser er målene for oxidative signaler til aktivering af NF-kB.140 Reduktionsmidler øger NF-kB DNA-binding, hvorimod oxidationsmidler hæmmer DNA-binding af NF-kB. TRX kan udøve 2 modsatrettede handlinger ved regulering af NF-kB: i cytoplasmaet blokerer det nedbrydning af IkB og hæmmer NF-kB-aktivering, men øger NF-kB DNA-binding i kernen.141 Aktivering af NF-kB via oxidationsrelateret nedbrydning af IkB resulterer i aktivering af adskillige antioxidantforsvarsrelaterede gener. NF-kB regulerer ekspressionen af flere gener, der deltager i immunrespons, såsom IL-1b, IL-6, tumornekrosefaktor-a, IL-8 og adskillige adhæsionsmolekyler.142,143 NF-kB regulerer også angiogenese og proliferation og differentiering af celler.
AP-1 er også reguleret af redoxtilstand. I nærvær af H2O2 kan nogle metalioner inducere aktivering af AP-1. Forøgelse i forholdet mellem GSH/GSSG øger AP-1-binding, mens GSSG hæmmer DNA-bindingen af AP-1.144. DNA-binding af Fos/Jun-heterodimeren øges ved reduktion af et enkelt konserveret cystein i det DNA-bindende domæne af hver af proteinerne,145 mens DNA-binding af AP-1 kan hæmmes af GSSG i mange celletyper, hvilket tyder på, at disulfidbindingsdannelse af cysteinrester hæmmer AP-1 DNA-binding.146,147 Signaltransduktion via oxidativ stress er opsummeret i figur 2.
KONKLUSIONER
Oxidativt stress kan opstå fra overproduktion af ROS ved metaboliske reaktioner, der bruger ilt og flytter balancen mellem oxidant/antioxidant status til fordel for oxidanterne. ROS produceres af cellulære metaboliske aktiviteter og miljøfaktorer, såsom luftforurenende stoffer eller cigaretrøg. ROS er meget reaktive molekyler på grund af uparrede elektroner i deres struktur og reagerer med flere biologiske makromolekyler i cellen, såsom kulhydrater, nukleinsyrer, lipider og proteiner, og ændrer deres funktioner. ROS påvirker også ekspressionen af flere gener ved opregulering af redox-følsomme transkriptionsfaktorer og kromatin-remodellering via ændring i histonacetylering/deacetylering. Regulering af redoxtilstand er kritisk for cellelevedygtighed, aktivering, proliferation og organfunktion.
REFERENCER
1. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Frie radikaler, metaller og antioxidanter i oxidativ stress-induceret cancer. Chem Biol Interact. 2006;160:1�40.
2. Halliwell B, Gutteridge JMC. Frie radikaler i biologi og medicin. 3. udg. New York: Oxford University Press;1999.
3. Marnett LJ. Lipidperoxidation-DNA-skade ved malondialdehyd. Mutat Res. 1999;424:83�95.
4. Siems WG, Grune T, Esterbauer H. 4-Hydroxynonenal dannelse under iskæmi og reperfusion af rotte tyndtarm. �Life Sci. 1995;57:785-789.
5. Stadtman ER. Oxiderende arters rolle i aldring. Curr Med Chem. 2004;11:1105�1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Lipidperoxidationsinducerede formodede malondialdehyd-DNA-addukter i humant brystvæv. Cancer Epidemiol Biomarkers Prev. 1996;5:705-710.
7. Jenner P. Oxidativ stress i Parkinsons sygdom. Ann Neurol. 2003;53: S26�S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. En vurdering af oxidativ skade på proteiner, lipider og DNA i hjernen fra patienter med Alzheimers sygdom. J Neurochem. 1997;68:2061�2069.
9. Sayre LM, Smith MA, Perry G. Kemi og biokemi af oxidativ stress i neurodegenerativ sygdom. Curr Med Chem. 2001;8:721�738.
10. Toshniwal PK, Zarling EJ. Beviser for øget lipidperoxidation ved multipel sklerose. Neurochem Res. 1992;17:205-207.
11. Dhalla NS, Temsah RM, Netticadan T. Rolle af oxidativ stress i hjerte-kar-sygdomme. J Hypertens. 2000;18:655�673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Undersøgelse af oxidativ stress i en rottemodel af kronisk hjernehyperfusion. Neurochem Int. 2005;46:601�611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxidanionproduktion øges i en model for genetisk hypertension: endotelets rolle. Forhøjet blodtryk. 1999;33:1353-1358.
14. Kukreja RC, Hess ML. Oxygen fri-radikal-systemet: fra ligninger gennem membran-protein-interaktioner til kardiovaskulær skade og beskyttelse. Cardiovasc Res. 1992;26:641-655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. Cigaretrygning inducerer en stigning i oxidativ DNA-skade, 8-hydroxydeoxyguanosin, på et centralt sted i den menneskelige lunge. Carcinogenese. 1997;18:1763-1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative og nitrosative hændelser ved astma. Free Radic Biol Med. 2003;35:213�225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Korrelation mellem systemisk superoxiddismutase-mangel og luftstrømsobstruktion ved astma. Am J Respir Crit Care Med. 2005;172:306�313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Superoxiddismutase-inaktivering i patofysiologi af astmatisk luftvejsremodellering og reaktivitet. Am J Pathol. 2005;166:663�674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Oxidativ stress og dets determinanter i luftvejene hos børn med astma. Allergi. 2008;63:1605�1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, et al. Oxidativt stress og genetiske og epidemiologiske determinanter for oxidantskade ved astma hos børn. J Allergy Clin Immunol. 2006;118:1097�1104.
21. Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Forskningsprogram for svær astma. Airway glutathion homeostase er ændret hos børn med svær astma: bevis for oxidant stress. J Allergy Clin Immunol. 2009;123:146�152.
22. Miller DM, Buettner GR, Aust SD. Overgangsmetaller som katalysatorer for "autooxidations"-reaktioner. Free Radic Biol Med. 1990;8:95�108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, D�me D, Pommier J. Mekanisme for hydrogenperoxiddannelse katalyseret af NADPH-oxidase i skjoldbruskkirtelplasmamembranen. J Biol Chem. 1991;266:3739-3743.
24. Granger DN. Rolle af xanthinoxidase og granulocytter i iskæmi-perfusionsskade. Am J Physiol. 1988;255:H1269�H1275.
25. Fenton HJH. Oxidation af vinsyre i nærvær af jern. J Chem Soc. 1984;65:899�910.
26. Haber F, Weiss JJ. Den katalytiske nedbrydning af hydrogenperoxid med jernsalte. Proc R Soc Lond Ser A. 1934;147:332�351.
27. Liochev SI, Fridovich I. The Haber�Weiss cyklede 70 år senere: et alternativt synspunkt. Redox Rep. 2002;7:55�57.
28. Klebanoff SJ. Myeloperoxidase: ven og fjende. J Leukoc Biol. 2005;77:598�625.
29. Whiteman M, Jenner A, Halliwell B. Hypochlorsyre-inducerede basemodifikationer i isoleret kalvethymus-DNA. Chem Res Toxicol. 1997;10:1240-1246.
30. Kulcharyk PA, Heinecke JW. Hypoklorsyre produceret af myeloperoxidasesystemet af humane fagocytter inducerer kovalente tværbindinger mellem DNA og protein. Biokemi. 2001;40:3648�3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, et al. En fortælling om to kontroverser: at definere både rollen af peroxidaser i nitrotyrosindannelse in vivo ved brug af eosinofil peroxidase og myeloperoxidasedeficiente mus, og arten af peroxidase-genererede reaktive nitrogenarter. J Biol Chem. 2002;277:17415�17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. Omfattende eosinofil degranulering og peroxidase-medieret oxidation af luftvejsproteiner forekommer ikke i en muse-ovalbumin-udfordringsmodel for lungebetændelse. J Immunol. 2001;167:1672-1682.
33. van Dalen CJ, Winterbourn CC, Senthilmohan R, Kettle AJ. Nitrit som substrat og inhibitor af myeloperoxidase. Implikationer for nitrering og hypoklorsyreproduktion på steder med inflammation. J Biol Chem. 2000;275:11638�11644.
34. Wood LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. Lipidperoxidation som bestemt af plasmaisoprostaner er relateret til sygdommens sværhedsgrad ved mild astma. Lipider. 2000;35:967�974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Øget 8-isoprostan, en markør for oxidativt stress, i udåndet kondensat fra astmapatienter. Am J Respir Crit Care Med. 1999;160:216�220.
36. Kirke DF, Pryor WA. Fri-radikal kemi af cigaretrøg og dens toksikologiske implikationer. Miljøsundhedsperspektiv. 1985;64:111-126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Ozon-induceret inflammation vurderet i sputum og bronkial lavagevæske fra astmatikere: et nyt ikke-invasivt værktøj i epidemiologiske undersøgelser af luftforurening og astma. Free Radic Biol Med. 1999;27:1448-1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Effekt af indåndet ozon på udåndet nitrogenoxid, lungefunktion og induceret opspyt hos normale og astmatiske personer. Thorax. 1999;54:1061-1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Redox-aktivitet af luftbårne partikler på forskellige steder i Los Angeles-bassinet. Environ Res. 2005;99:40�47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Differentiel induktion af ekstracellulær glutathionperoxidase og nitrogenoxidsyntase 2 i luftvejene hos raske individer udsat for 100 % O(2) eller cigaretrøg. Am J Respir Cell Mol Biol. 2000;23:350�354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Oxidant-medieret lungeskade i akut respiratorisk distress syndrom. Crit Care Med. 1999;27:2028-2030.
42. Biaglow JE, Mitchell JB, Held K. Vigtigheden af peroxid og superoxid i røntgenresponsen. Int J Radiat Oncol Biol Phys. 1992;22:665-669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Kobberion-medieret sensibilisering af nuklear matrix-bindingssteder over for ioniserende stråling. Biokemi. 1993;32:6214�6219.
44. Narayanan PK, Goodwin EH, Lehnert BE. Alfa-partikler initierer biologisk produktion af superoxidanioner og hydrogenperoxid i humane celler. Cancer Res. 1997;57:3963�3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Følsomhed over for kemiske oxidanter og stråling i CHO-cellelinjer, der mangler oxidativ pentosecyklusaktivitet. Int J Radiat Oncol Biol Phys. 1992;22: 671�675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, et al. Mangan
superoxiddismutase-medieret genekspression i strålingsinduceret
adaptive reaktioner. Mol Cell Biol. 2003;23:2362�2378.
47. Azzam EI, de Toledo SM, Spitz DR, Lille JB. Oxidativ metabolisme
modulerer signaltransduktion og dannelse af mikrokerner hos tilstedeværende
celler fra a-partikel-bestrålede normale humane fibroblaster. Cancer Res.
2002; 62: 5436 5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Ioniserende stråling-induceret, mitokondrier-afhængig generering af reaktive
ilt/nitrogen. Cancer Res. 2001;61:3894�3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK-veje i
strålingsreaktioner. Onkogen. 2003;22:5885�5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, et al. Thioredoxin
nuklear translokation og interaktion med redoxfaktor-1 aktiverer AP-1-transkriptionsfaktoren som reaktion på ioniserende stråling. Cancer Res. 2000;60:6688�6695.
51. Cadet J, Douki T, Gasparutto D, Ravanat JL. Oxidativ skade på DNA: dannelse, måling og biokemiske egenskaber. Mutat Res. 2003;531:5�23.
52. Yokoya A, Cunniffe SM, O�Neill P. Effekt af hydrering på induktion af strengbrud og baselæsioner i plasmid-DNA-film ved gammastråling. J Am Chem Soc. 2002;124:8859�8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Celle- og vævsreaktioner på oxidativ skade. Lab Invest. 1993;69:261-274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Nuklear faktor kappa B-afhængig induktion af gamma-glutamylcysteinsyntetase ved ioniserende stråling i T98G humane glioblastomceller. Free Radic Biol Med. 1998;24:1256-1268.
55. Stohs SJ, Bagchi D. Oxidative mekanismer i toksiciteten af metalioner. Free Radic Biol Med. 1995;18:321-336.
56. Leonard SS, Harris GK, Shi X. Metal-induceret oxidativ stress og signaltransduktion. Free Radic Biol Med. 2004;37:1921-1942.
57. Shi H, Shi X, Liu KJ. Oxidativ mekanisme for arsen toksicitet og carcinogenese. Mol Cell Biochem. 2004;255:67�78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. En potentiel mekanisme for forringelse af nitrogenoxiddannelse forårsaget af langvarig oral eksponering for arsenat hos kaniner. Free Radic Biol Med.2003;35:102�113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. DNA-strengbrud induceret af dimethylarsinsyre, en metabolit af uorganisk arsenik, forstærkes kraftigt af superoxidanionradikaler. Biol Pharm Bull. 1995;18:45�58.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Mekanismer, der ligger til grund for arsencarcinogenese: overfølsomhed hos mus udsat for uorganisk arsen under drægtighed. Toksikologi. 2004;198:31�38.
61. Schiller CM, Fowler BA, Woods JS. Effekter af arsen på aktivering af pyruvatdehydrogenase. Miljøsundhedsperspektiv. 1977;19:205-207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Frie radikalers involvering i neurologiske porfyrier og blyforgiftning. Mol Cell Biochem. 1991;103:73-83.
63. Tripathi RM, Raghunath R, Mahapatra S. Blodbly og dets virkning på Cd, Cu, Zn, Fe og hæmoglobinniveauer hos børn. Sci Total Environ. 2001;277:161-168.
64. Nehru B, Dua R. Effekten af kostselen på blyneurotoksicitet. J Environ Pathol Toxicol Oncol. 1997;16:47�50.
65. Reid TM, Feig DI, Loeb LA. Mutagenese af metal-inducerede oxygenradikaler. Miljøsundhedsperspektiv. 1994;102(suppl 3):57�61.
66. Kinnula VL, Crapo JD. Superoxiddismutaser i lungerne og menneskelige lungesygdomme. Am J Respir Crit Care Med. 2003;167:1600–1619.
67. Kinnula VL. Produktion og nedbrydning af oxygenmetabolitter under inflammatoriske tilstande i den menneskelige lunge. Curr Drug Targets Inflamm Allergi. 2005;4:465�470.
68. Zelko IN, Mariani TJ, Folz RJ. Superoxiddismutase multigenfamilie: en sammenligning af CuZn-SOD (SOD1), Mn-SOD (SOD2) og EC-SOD (SOD3) genstrukturer, evolution og ekspression. Free Radic Biol Med. 2002;33:337�349.
69. Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Mekanismer til beskyttelse af katalase ved NADPH. Kinetik og støkiometri. J Biol Chem. 1999;274:13908�13914.
70. Floh� L. Glutathionperoxidase. Basic Life Sci. 1988;49:663-668.
71. Arthur JR. Glutathionperoxidaserne. Cell Mol Life Sci. 2000;57:1825-1835.
72. Chu FF, Doroshow JH, Esworthy RS. Ekspression, karakterisering og vævsfordeling af en ny cellulær selen-afhængig glutathionperoxidase, GSHPx-GI. J Biol Chem. 1993;268:2571-2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Ekstracellulær glutathionperoxidase-induktion i astmatiske lunger: bevis for redoxregulering af ekspression i humane luftvejsepitelceller. FASEB J. 2001;15:70�78.
74. Gromer S, Urig S, Becker K. Thioredoxinsystemet fra videnskab til klinik. Med Res Rev. 2004;24:40�89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, P��kk� P, et al. Cellespecifik ekspression af peroxiredoxiner i human lunge- og pulmonal sarkoidose. Thorax. 2002;57:157�164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T, et al. Humant peroxiredoxin 5 er en peroxynitritreduktase. FEBS Lett. 2004;571:161�165.
77. Holmgren A. Antioxidantfunktion af thioredoxin og glutaredoxinsystemer. Antioxid redox signal. 2000;2:811�820.
78. Dickinson DA, Forman HJ. Glutathion i forsvar og signalering: lektioner fra en lille thiol. Ann NY Acad Sci. 2002;973:488�504.
79. Sies H. Glutathion og dets rolle i cellulære funktioner. Free Radic Biol Med. 1999;27:916-921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Parallelle evolutionære veje for glutathiontransferaser: struktur og mekanisme af mitokondrieklassen kappa-enzymet rGSTK1-1. Biokemi. 2004;43:52�61.
81. Robinson A, Huttley GA, Booth HS, Board PG. Modellerings- og bioinformatikundersøgelser af den humane kappa-klasse glutathiontransferase forudsiger en ny tredje transferasefamilie med homologi til prokaryote 2-hydroxychromene-2-carboxylat-isomeraser. Biochem J. 2004;379:541-552.
82. Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Fælles strukturelle træk ved MAPEGda udbredte superfamilie af membranassocierede proteiner med meget divergerende funktioner i eicosanoid- og glutathionmetabolisme. Protein Sci. 1999;8:689-692.
83. Hayes JD, Pulford DJ. Glutathion S-transferase-supergenfamilien: regulering af GST og isoenzymes bidrag til cancerkemobeskyttelse og lægemiddelresistens. Crit Rev Biochem Mol Biol. 1995;30:445�600.
84. Armstrong RN. Struktur, katalytisk mekanisme og udvikling af glutathiontransferaser. Chem Res Toxicol. 1997;10:2�18.
85. Hayes JD, McLellan LI. Glutathion og glutathion-afhængige enzymer repræsenterer et koordineret reguleret forsvar mod oxidativt stress. Free Radic Res. 1999;31:273�300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Struktur, funktion og udvikling af glutathiontransferaser: implikationer for klassificering af ikke-pattedyrsmedlemmer af en gammel enzymsuperfamilie. Biochem J. 2001;360:1-16.
87. Cho SG, Lee YH, Park HS, Ryoo K, Kang KW, et al. Glutathion S-transferase Mu modulerer de stressaktiverede signaler ved at undertrykke apoptose signalregulerende kinase 1. J Biol Chem. 2001;276:12749�12755.
88. Dorion S, Lambert H, Landry J. Aktivering af p38-signalvejen ved varmechok involverer dissocieringen af glutathion S-transferase Mu fra Ask1. J Biol Chem. 2002;277:30792�30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, et al. Regulering af JNK-signalering ved GSTp. EMBO J. 1999;18:1321-1334.
90. Manevich Y, Feinstein SI, Fisher AB. Aktivering af antioxidantenzymet 1-CYS peroxiredoxin kræver glutathionylering medieret af heterodimerisering med pGST. Proc Natl Acad Sci US A. 2004;101:3780�3785.
91. Bunker VW. Frie radikaler, antioxidanter og aldring. Med Lab Sci. 1992;49:299�312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD, et al. Systemisk oxidativ stress og dets sammenhæng med alder og sygdom. J Am Geriatr Soc. 1996;44:823�827.
93. White E, Shannon JS, Patterson RE. Forholdet mellem vitamin og
brug af calciumtilskud og tyktarmskræft. Cancer Epidemiol Biomarkers Prev. 1997;6:769-774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Nye mekanismer af naturlige antioxidantforbindelser i biologiske systemer: involvering af glutathion og glutathion-relaterede enzymer. J Nutr Biochem. 2005;16:577�586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Ændringer i hjertets glutathionstatus efter iskæmi og reperfusion. Experientia. 1985;41:42�43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Carotenoid radikal kemi og antioxidant/pro-oxidant egenskaber. Arch Biochem Biophys. 2004;430:37�48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Hvorfor forventer vi, at carotenoider er antioxidanter in vivo? Free Radic Res. 1997;26:381-398.
98. Nils RM. Signalveje i retinoid kemoforebyggelse og behandling af cancer. Mutat Res. 2004;555:81�96.
99. Donato LJ, Noy N. Undertrykkelse af brystkarcinomvækst af retinsyre: proapoptotiske gener er mål for retinsyrereceptor og cellulær retinsyrebindende protein II-signalering. Cancer Res. 2005;65:8193�8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. Bcl-2 er en nøgleregulator for retinsyre-induceret apoptotisk celledød i neuroblastom. Onkogen. 2006;25:5046�5055.
101. Dalton TP, Shetzer HG, Puga A. Regulering af genekspression ved hjælp af reaktivt oxygen. Ann Rev Pharmacol Toxicol. 1999;39:67�101.
102. Scandalios JG. Genomiske reaktioner på oxidativt stress. I: Meyers RA, red. Encyclopedia of Molecular Cell Biology and Molecular Medicine. bind 5. 2. udg. Weinheim, Tyskland: Wiley-VCH; 2004: 489�512.
103. Ghosh R, Mitchell DL. Effekt af oxidativ DNA-skade i promotorelementer på transkriptionsfaktorbinding. Nucleic Acids Res. 1999;27:3213�3218.
104. Marietta C, Gulam H, Brooks PJ. En enkelt 8-cyclo-50-deoxyadenosin-læsion i en TATA-boks forhindrer binding af det TATA-bindende protein og reducerer kraftigt transkription in vivo. DNA Reparation (Amst). 20;2002:1�967.
105. Jackson AL, Chen R, Loeb LA. Induktion af mikrosatellit-ustabilitet
ved oxidativ DNA-skade. Proc Natl Acad Sci US A. 1998;95:12468�12473.
106. Caldecott KW. Protein-protein-interaktioner under reparation af pattedyr-DNA-enkeltstrengsbrud. Biochem Soc Trans. 2003;31:247�251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidativ DNA-skade: mekanismer, mutation og sygdom. FASEB J. 2003;17:1195�1214.
108. Jones PL, Wolffe AP. Relationer mellem kromatinorganisation og DNA-methylering ved bestemmelse af genekspression. Semin Cancer Biol. 1999;9:339-347.
109. Girotti AW. Mekanismer for lipidperoxidation. J Free Radic Biol Med. 1985;1:87�95.
110. Siu GM, Draper HH. Metabolisme af malonaldehyd in vivo og in vitro. Lipider. 1982;17:349-355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Mulig involvering af lipid-peroxidationsproduktet 4-hydroxynonenal i dannelsen af fluorescerende kromolipider. Biochem J. 1986;239:405-409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Aldersafhængige ændringer i lipidperoxidniveauer i lipoproteinfraktionerne af humant serum. J Gerontol. 1984;39:269-272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- Hydroxynonenal, et aldehydisk produkt af membranlipidperoxidation, forringer glutamattransport og mitokondriel funktion i synaptosomer. Neurovidenskab. 1997;806:85�96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Aktivering af stresssignalveje ved slutproduktet af lipidperoxidation. 4-hydroxy-2-nonenal er en potentiel inducer af intracellulær peroxidproduktion. J Biol Chem. 1999;274:2234�2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Aktivering af EGF-receptor ved oxideret LDL. FASEB J. 1998;12:665�671.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal øger fibronectinproduktion af IMR-90 humane lungefibroblaster delvist via aktivering af epidermal vækstfaktor receptor-bundet ekstracellulært signal- reguleret kinase p44/42 pathway. Toxicol Appl Pharmacol. 2002;184:127-135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Udåndet 8-isoprostan som en in vivo biomarkør for lungeoxidativt stress hos patienter med KOL og raske rygere. Am J Respir Crit Care Med. 2000;162:1175-1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Epitelpermeabilitet, inflammation og oxidant stress i rygeres luftrum. Am J Respir Crit Care Med. 1999;159:473-479.
119. Nowak D, Kasielski M, Antczak A, Pietras T, Bialasiewicz P. Øget indhold af thiobarbitursyre-reaktive stoffer og hydrogenperoxid i udåndingskondensatet hos patienter med stabil kronisk obstruktiv lungesygdom: ingen signifikant effekt af cigaretrygning. Respir Med. 1999;93:389-396.
120. Kelly FJ, Mudway IS. Proteinoxidation ved luft-lunge-grænsefladen. Aminosyrer. 2003;25:375�396.
121. Dean RT, Roberts CR, Jessup W. Fragmentering af ekstracellulære og intracellulære polypeptider af frie radikaler. Prog Clin Biol Res. 1985;180:341-350.
122. Keck RG. Anvendelsen af t-butylhydroperoxid som en sonde til methioninoxidation i proteiner. Anal Biochem. 1996;236:56-62.
123. Davies KJ. Proteinskade og nedbrydning af iltradikaler. I. Generelle aspekter. J Biol Chem. 1987;262:9895-9901.
124. Stadtman ER. Metalion-katalyseret oxidation af proteiner: biokemisk mekanisme og biologiske konsekvenser. Free Radic Biol Med.
1990; 9: 315 325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inaktivering af vigtige metaboliske enzymer ved blandede oxidationsreaktioner: mulig implikation i proteinomsætning og aldring. Proc Natl Acad Sci US A. 1983;80:1521�1525.
126. Stadtman ER, Moskovitz J, Levine RL. Oxidation af methioninrester af proteiner: biologiske konsekvenser. Antioxid redox signal. 2003;5:577�582.
127. Stadtman ER, Levine RL. Fri radikal-medieret oxidation af frie aminosyrer og aminosyrerester i proteiner. Aminosyrer. 2003;25:207�218.
128. Stadtman ER. Proteinoxidation ved aldring og aldersrelaterede sygdomme. Ann NY Acad Sci. 2001;928:22�38.
129. Shacter E. Kvantificering og betydning af proteinoxidation i biologiske prøver. Drug Metab Rev. 2000;32:307�326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidativ stress og cellesignalering. Curr Med Chem. 2004;11:1163-1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vaskulær endotelvækstfaktor (VEGF) og dens receptorer. FASEB J. 1999;13:9�22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulering af generering af reaktive iltarter i fibroblaster af Rac1. Biochem J. 1996;318:379-382.
133. Sun T, Oberley LW. Redox-regulering af transkriptionelle aktivatorer. Free Radic Biol Med. 1996;21:335-348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox-regulering af c-Jun DNA-binding ved reversibel S-glutathiolation. FASEB J. 1999;13:1481-1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. In situ påvisning af S-glutathionylerede proteiner efter glutaredoxin-1-katalyseret cysteinderivatisering. Biochim Biophys Acta. 2006;1760:380�387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation af glutaredoxin-1
udtryk i en musemodel af allergisk luftvejssygdom. Am J Respir Cell Mol Biol. 2007;36:147�151.
137. Filomeni G, Rotilio G, Ciriolo MR. Cellesignalering og glutathion-redox-systemet. Biochem Pharmacol. 2002;64:1057-1064.
138. Pande V, Ramos MJ. Molekylær genkendelse af 15-deoxydelta (12,14) prostaglandin J(2) af nuklear faktor-kappa B og andre cellulære proteiner. Bioorg Med Chem Lett. 2005;15:4057�4063.
139. Perkins ND. Integrering af cellesignaleringsveje med NF-kappaB og IKK funktion. Nat Rev Mol Cell Biol. 2007;8:49�62.
140. Gilmore TD. Introduktion til NF-kappaB: spillere, veje, perspektiver. Onkogen. 2006;25:6680�6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinkte roller af thioredoxin i cytoplasmaet og i kernen. En to-trins mekanisme til redoxregulering af transkriptionsfaktor NF-kappaB. J Biol Chem. 1999;274:27891�27897.
142. Afdeling PA. Rolle af komplement, kemokiner og regulatoriske cytokiner ved akut lungeskade. Ann NY Acad Sci. 1996;796:104�112.
143. Akira S, Kishimoto A. NF-IL6 og NF-kB i cytokin-genregulering. Adv Immunol. 1997;65:1�46.
144. Meyer M, Schreck R, Baeuerle PA. H2O2 og antioxidanter har modsatte virkninger på aktivering af NF-kappa B og AP-1 i intakte celler: AP-1 som sekundær antioxidant-responsiv faktor. EMBO J. 1993;12:2005�2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Redox-regulering af fos- og jun-DNA-bindende aktivitet in vitro. Videnskab. 1990;249:1157-1161.
146. Galter D, Mihm S, Droge W. Distinkte virkninger af glutathiondisulfid på de nukleare transkriptionsfaktorer kB og aktivatorproteinet-1. Eur J Biochem. 1994;221:639-648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. AP-1 transkriptionel aktivitet er reguleret af en direkte association mellem thioredoxin og Ref-1. Proc Natl Acad Sci US A. 1997;94: 3633�3638.
IFM's Find A Practitioner-værktøj er det største henvisningsnetværk inden for Functional Medicine, skabt for at hjælpe patienter med at finde Functional Medicine-praktikere overalt i verden. IFM Certified Practitioners er anført først i søgeresultaterne på grund af deres omfattende uddannelse i Functional Medicine