
Neuroinflammation:
Abstrakt
Flere bevislinjer understøtter den patogene rolle af neuroinflammation i psykiatrisk sygdom. Mens systemiske autoimmune sygdomme er veldokumenterede årsager til neuropsykiatriske lidelser, bliver synaptiske autoimmune encephalitider med psykotiske symptomer ofte underkendt. Parallelt med sammenhængen mellem psykiatriske symptomer og autoimmunitet i autoimmune sygdomme, forekommer neuroimmunologiske abnormiteter ved klassiske psykiatriske lidelser (f.eks. svær depressiv, bipolar, skizofreni og tvangslidelser). Undersøgelser af patofysiologien af disse tilstande understregede traditionelt dysregulering af de glutamaterge og monoaminerge systemer, men mekanismerne, der forårsagede disse neurotransmitterabnormaliteter, forblev uhåndgribelige. Vi gennemgår sammenhængen mellem autoimmunitet og neuropsykiatriske lidelser og de menneskelige og eksperimentelle beviser, der understøtter den patogene rolle af neuroinflammation i udvalgte klassiske psykiatriske lidelser. At forstå, hvordan psykosociale, genetiske, immunologiske og neurotransmittersystemer interagerer, kan afsløre patogene spor og hjælpe med at målrette mod nye forebyggende og symptomatiske terapier.
nøgleord:
- Neuroinflammation,
- Psykoneuroimmunologi,
- Astrocyt,
- Microglia,
- Cytokiner,
- Oxidativt stress,
- Depression,
- Tvangslidelse,
- Bipolar lidelse, skizofreni
Indhold
Introduktion
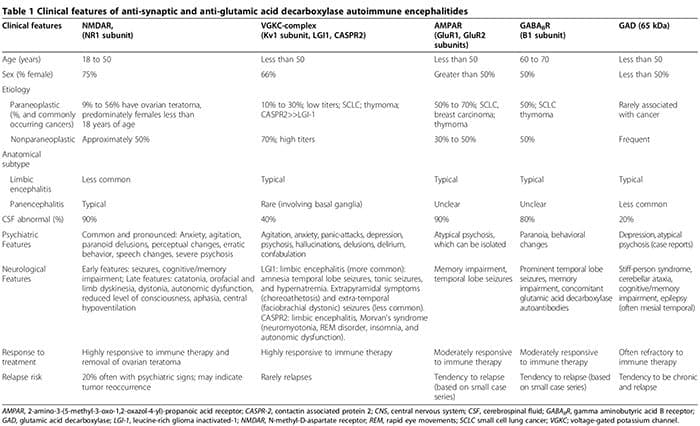
Efterhånden som biologiske abnormiteter i stigende grad identificeres blandt patienter med psykiatriske lidelser, forsvinder sondringen mellem neurologisk og psykiatrisk sygdom. Ud over systemiske autoimmune sygdomme forbundet med psykiatriske manifestationer (for eksempel lupus) [1], blev patienter med akut isoleret psykose for nylig identificeret med synaptiske autoimmune encephalitider (tabel 1) [2-6]. Disse patienter er ofte fejlagtigt diagnosticeret med refraktære primære psykotiske lidelser, hvilket forsinker påbegyndelsen af effektiv immunterapi (tabel 1). Derudover understøtter voksende beviser den patogene rolle af anti-neuronale antistoffer i neuropsykiatriske lidelser [7].
Adskillelse af neurologiske og psykiatriske lidelser, understøttet af Descartes' opfattelse af �sindet� som en ontologisk distinkt enhed og af reproducerbarheden af neuropatologiske abnormiteter, dominerede medicin i�det 19. og det tidlige 20. århundrede [8]. Siden da har en voksende samling af reproducerbare biologiske årsager, fra neurosyfilis, hovedtraume, slagtilfælde, tumor, demyelinisering og mange andre forårsaget symptomkomplekser, der overlappede med klassiske psykiatriske lidelser [9-11]. For nylig er neuroinflammatoriske og immunologiske abnormiteter blevet dokumenteret hos patienter med klassiske psykiatriske lidelser.
Perifere immunmodulatorer kan inducere psykiatriske symptomer i dyremodeller og mennesker [12-19]. Raske dyr injiceret med pro-inflammatorisk IL-1? og tumornekrosefaktor alfa (TNF-?) cytokiner viser "sygdomsadfærd" forbundet med social tilbagetrækning [12]. Hos mennesker deaktiverer injektioner af lavdosis endotoksin det ventrale striatum, et område, der er afgørende for belønningsbehandling, hvilket producerer anhedoni, et invaliderende depressivt symptom [14]. Cirka 45 % af ikke-deprimerede hepatitis C- og cancerpatienter behandlet med IFN-? udvikle depressive symptomer forbundet med øgede serum IL-6 niveauer [12,15,17,18].
Medicinske tilstande forbundet med kroniske inflammatoriske og immunologiske abnormiteter, herunder fedme, diabetes, maligniteter, rheumatoid arthritis og multipel sklerose, er risikofaktorer for depression og bipolar lidelse [10,12,13,15,17,18]. Det positive�sammenhæng mellem disse medicinske tilstande og psykiatrisk sygdom tyder på tilstedeværelsen af en udbredt underliggende inflammatorisk proces, der påvirker hjernen blandt andre organer [10,19,20]. En 30-årig befolkningsbaseret undersøgelse viste, at have en autoimmun sygdom eller en tidligere indlæggelse for alvorlig infektion øgede risikoen for at udvikle skizofreni med henholdsvis 29 % og 60 % [16]. Herpes simplex-virus, Toxoplasma gondii, cytomegalovirus og influenza under graviditet øger desuden risikoen for at udvikle skizofreni [16].
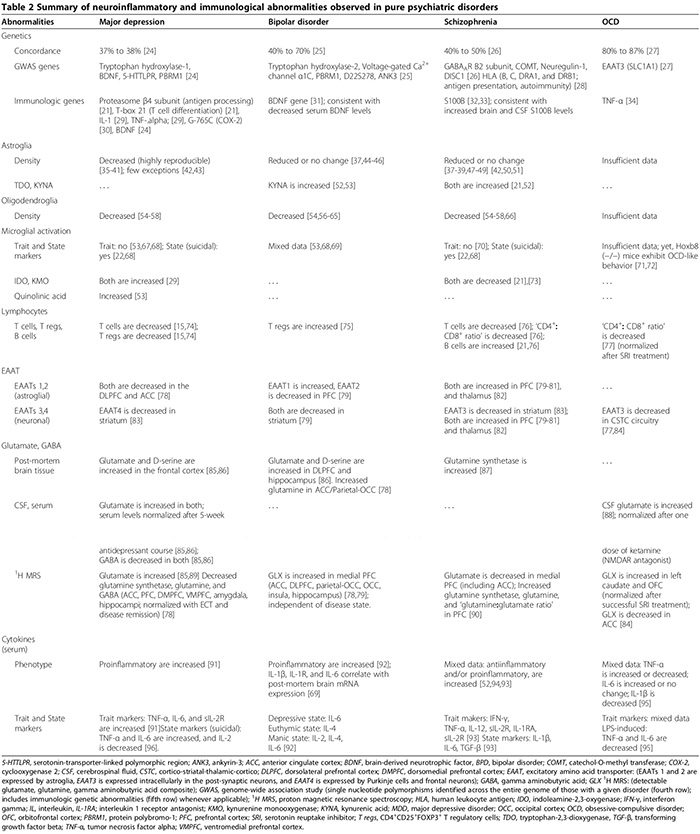
Perifere cellulære [21,22] (tabel 2) og humorale immunologiske abnormiteter [13,21-23] er mere udbredt hos psykiatriske patienter i forhold til raske kontroller. I både pilot- (n = 34 patienter med svær depressiv lidelse (MDD), n = 43 raske kontroller) og replikationsundersøgelser (n = 36 MDD, n = 43 raske kontroller), adskilte et serumassay, der omfattede ni serumbiomarkører, MDD-personer fra raske kontroller med 91.7 % sensitivitet og 81.3 %. signifikant forhøjede biomarkører for neuropsykiatriske symptomer var de immunologiske molekyler alfa 1 antitrypsin, myeloperoxidase og opløselig TNF-? receptor II [23].
 Vi gennemgår først sammenhængen mellem autoimmunitet og neuropsykiatriske lidelser, herunder: 1) systemisk lupus erythematosus (SLE) som en prototype af systemisk autoimmun sygdom; 2) autoimmune encephalitider forbundet med serum anti-synaptiske og glutaminsyre decarboxylase (GAD) autoantistoffer; og 3) pædiatriske neuropsykiatriske autoimmune lidelser forbundet med streptokokinfektioner (PANDAS) og ren obsessiv-kompulsiv lidelse (OCD) forbundet med anti-basale ganglier/thalamus autoantistoffer. Vi diskuterer derefter rollen af medfødt inflammation/autoimmunitet i klassiske psykiatriske lidelser, herunder MDD, bipolar lidelse (BPD), skizofreni og OCD.
Vi gennemgår først sammenhængen mellem autoimmunitet og neuropsykiatriske lidelser, herunder: 1) systemisk lupus erythematosus (SLE) som en prototype af systemisk autoimmun sygdom; 2) autoimmune encephalitider forbundet med serum anti-synaptiske og glutaminsyre decarboxylase (GAD) autoantistoffer; og 3) pædiatriske neuropsykiatriske autoimmune lidelser forbundet med streptokokinfektioner (PANDAS) og ren obsessiv-kompulsiv lidelse (OCD) forbundet med anti-basale ganglier/thalamus autoantistoffer. Vi diskuterer derefter rollen af medfødt inflammation/autoimmunitet i klassiske psykiatriske lidelser, herunder MDD, bipolar lidelse (BPD), skizofreni og OCD.
Neuropsykiatriske lidelser forbundet med autoimmunitet
Systemisk lupus erythematosus
Mellem 25 % og 75 % af SLE-patienter har centralnervesystemet (CNS) involvering, med psykiatriske symptomer typisk inden for de første to år efter sygdomsdebut. Psykiatriske symptomer kan omfatte angst, humør og psykotiske forstyrrelser [97]. Hjernemagnetisk resonansbilleddannelse (MRI) er normal i cirka 42 % af neuropsykiatriske SLE-tilfælde [97]. Mikroangiopati og nedbrydning af blodhjernebarrieren (BBB) kan tillade, at autoantistoffer trænger ind i hjernen [97]. Disse antistoffer omfatter anti-ribosomal P (positiv hos 90% af psykotiske SLE-patienter) [1], anti-endotelcelle, anti-gangliosid, anti-dsDNA, anti-2A/2B underenheder af N-methyl-D-aspartatreceptorer (NMDAR) og anti-phospholipid antistoffer [97]. Pro-inflammatoriske cytokiner�hovedsagelig IL-6 [97], S100B�[97], intracellulært adhæsionsmolekyle 1 [97] og matrix-metalloproteinase-9 [98] er også forhøjet i SLE. Psykiatriske manifestationer af SLE, Sjo?grens sygdom, Susacs syndrom, CNS vaskulitis, CNS Whipples sygdom og Behc?et's sygdom blev for nylig gennemgået [1].
Neuropsykiatriske autoimmune encephalitider associeret med serum anti-synaptisk og glutaminsyre decarboxylase
autoantistoffer
Autoimmune encephalitider er karakteriseret ved en akut indtræden af tindingelappens anfald, psykiatriske træk og kognitive mangler [2,3,99-108]. Patofysiologien er typisk medieret af autoantistoffer rettet mod synaptiske eller intracellulære autoantigener i forbindelse med en paraneo plastisk eller ikke-paraneoplastisk oprindelse [3]. Anti-synaptiske autoantistoffer retter sig mod NR1-underenheder af NMDAR [100,108,109], spændingsstyrede kaliumkanal- (VGKC)-komplekser (Kv1-underenhed, leucinrigt gliominaktiveret (LGI1) og kontaktinassocieret protein 2 (CASPR2)) [101,102,106-hydroxy-amino-gluR1, GluR2, 3, hydroxy] 5-methyl-l-4-isoxazolpropionsyrereceptor (AMPAR) [6,110,111] og B1-underenheder af β-aminosmørsyre B-receptorerne (GABABR) [3,99,103]. Anti-intracellulære autoantistoffer retter sig mod onconeuronale og GAD-65 autoantigener [2,3].
Inflammationen forbundet med anti-synaptiske autoantistoffer, især NMDAR-autoantistoffer, er typisk meget mildere end den, der er forbundet med GAD-autoantistoffer eller anti-neuronale autoantistoffer relateret til systemiske autoimmune lidelser eller paraneoplastiske syndromer [2,107].
Selvom neurologiske symptomer til sidst dukker op, kan psykiatriske manifestationer, lige fra angst [2,3] til psykose, der efterligner skizofreni [2-6], i starten dominere eller gå forud for neurologiske træk. Op til to tredjedele af patienterne med anti-NMDAR autoimmun encephalitis, er oprindeligt til stede på psykiatriske tjenester [5]. Anti-synaptiske antistof-medierede autoimmune encephalitider skal overvejes i differentialet af akut psykose [2-6]. Psykiatriske præsentationer kan omfatte normal hjerne-MRI og cerebrospinalvæske (CSF) analyse, uden encefalopati eller kramper [2,3,5,6,107]. Vi rapporterede et tilfælde af seropositive GAD-autoantistoffer forbundet med biopsi-bevist neuroinflammation, på trods af normal hjerne-MRI og CSF-analyser, hvor patienten præsenterede med isoleret psykose diagnosticeret som skizofreni af Diagnostic and Statistical Manual of Mental Disorders, 4. udgave (DSM-IV) kriterier [2]. Yderligere kan seronegative autoimmune encephalitider også præsentere sig med fremtrædende neuropsykiatriske forstyrrelser, hvilket gør diagnosen mere uhåndgribelig [107,112,113]. Psykiatriske og neurologiske træk forbundet med antisynaptiske og GAD-autoantistoffer er opsummeret i tabel 1 [1-6,99-108,114].
Serum anti-synaptiske og GAD autoantistoffer kan forekomme hos patienter med rene psykiatriske lidelser [2,4,5,112,115-121]. I en prospektiv kohorte på 29 forsøgspersoner, der opfyldte DSM-IV-kriterierne for skizofreni, blev der fundet serum-anti-NMDAR-autoantistoffer i tre forsøgspersoner, og anti-VGKC-komplekse autoantistoffer blev fundet i et individ [5]. Ved at bruge mere følsomme teknikker til at påvise immunoglobulin G (IgG) NR1-autoantistoffer hos 100 patienter med decideret skizofreni, blev der ikke identificeret nogen autoantistoffer [122]. Imidlertid vurderede denne undersøgelse ikke autoantistoffer rettet mod NR2-underenheden af NMDAR. Andre undersøgelser rapporterede signifikant øgede odds for forhøjede (?90. percentil ikke-psykiatriske kontrolniveauer) NR2-antistofniveauer (odds ratio (OR) 2.78, 95 % konfidensinterval (CI) 1.26 til 6.14, P = 0.012) blandt individer med akut mani (n = 43, men ikke inkronisk mani (n = 116]).
PANDAS & ren obsessiv-kompulsiv lidelse forbundet med anti-basale ganglier/thalamus autoantistoffer
OCD komplicerer ofte neurologiske lidelser, der involverer basalganglierne, herunder Sydenhams chorea, Huntingtons sygdom og Parkinsons sygdom. Anti-basale ganglier antistoffer er impliceret i Sydenhams chorea [123]. PANDAS er karakteriseret ved akutte eksacerbationer af OCD-symptomer og/eller motoriske/foniske tics efter en prodromal gruppe A β-hæmolytisk streptokokinfektion. Patofysiologien menes at involvere krydsreaktivitet mellem anti-streptokok-antistoffer og basale gangliaproteiner [124]. Det kliniske overlap mellem PANDAS og ren OCD antyder en fælles ætiologisk mekanisme [125].
Blandt en tilfældig kohorte på 21 rene OCD-patienter havde 91.3% CSF anti-basale ganglier (P <0.05) og anti-thalamiske autoantistoffer (P <0.005) ved 43 kDa [88], parallelt med funktionelle abnormiteter i cortico-striatal-thalamo-cortico-kredsløbene i OCD-subjektskredsløbet [84]. En anden undersøgelse dokumenterede, at 42 % (n = 21) af OCD pædiatriske og unge forsøgspersoner havde serum anti-basale ganglier autoantistoffer ved 40, 45 og 60 kDa sammenlignet med 2 % til 10 % af kontrollerne (P = 0.001) [7]. Anti-basale ganglia-autoantistoffer blev påvist i sera fra 64 % af PANDAS-individerne (n = 14), sammenlignet med kun 9 % (n = 2) af streptokok-positive/OCD-negative kontroller (P <0.001) [126]. En undersøgelse fandt ingen forskel mellem prævalensen af anti-basale ganglier autoantistoffer i OCD (5.4%, n = 4) versus MDD kontroller (0%) [127]; en begrænsning var imidlertid den tilfældige brug af rottecortex og bovine basale ganglier og cortex, som kunne have begrænset identifikation af seropositive tilfælde.
De basale ganglia-autoantigener er aldolase C (40 kDa), neuronspecifik/ikke-neuronal enolase (45 kDa dublet) og pyruvatkinase M1 (60 kDa) - neuronale glykolytiske enzymer - involveret i neurotransmission, neuronal metabolisme
Side 3 af 24 og cellesignalering [128]. Disse enzymer udviser væsentlig strukturel homologi med streptokokproteiner [129]. Den seneste undersøgelse (96 OCD, 33 MDD, 17 skizofreni-patienter) testede specifikt patientserum mod pyruvatkinase, aldolase C og enolase; en større andel af OCD-personer var seropositive i forhold til kontrollerne (19.8 % (n = 19) versus 4 % [n = 2], P = 0.012) [130].
Alligevel havde kun én af 19 seropositive OCD-personer i samme undersøgelse positive anti-streptolysin O-antistoftitre, hvilket tyder på, at i ren OCD udelukker anti-streptolysin O-antistofseronegativiteten ikke tilstedeværelsen af anti-basale ganglia-autoantistoffer.
Ved ren OCD er sero-positivitet for anti-basale ganglier/thalamus-antistoffer forbundet med øgede niveauer af CSF-glycin (P = 0.03) [88], hvilket tyder på, at disse antistoffer bidrager til hyperglutamatergi observeret i OCD [84,88,131]. Forbedringen af infektionsfremkaldt OCD med immunterapier understøtter disse autoantistoffers patogenicitet [132]. Et stort NIH-forsøg, der vurderer effektiviteten af intravenøst immunglobulin (IVIG) til børn med akut indsættende OCD og anti-streptokok-antistoffer er i gang (ClinicalTrials.gov: NCT01281969). Men fundet af lidt højere CSF-glutamatniveauer hos OCD-patienter med negative CSF anti-basale ganglier/thalamus-antistoffer sammenlignet med dem med positive CSF-antistoffer tyder på, at ikke-immunologiske mekanismer kan spille en rolle i OCD [84]. Andre mekanismer, herunder cytokin-medieret inflammation (tabel 2), er også hypotese.
Psykiatriske lidelser forbundet med medfødt betændelse
Lidelser af medfødt inflammation/autoimmunitet forekommer hos nogle patienter med klassiske psykiatriske lidelser. Vi diskuterer medfødte betændelsesrelaterede CNS-abnormiteter� inklusive gliapatologi, forhøjede cytokinniveauer, cyclooxygenaseaktivering, glutamatdysregulering, øgede S100B-niveauer, øget oxidativt stress og BBB-dysfunktion�i MDD, BPD, skizofreni og OCD. Vi beskriver også, hvordan medfødt inflammation kan være mekanisk forbundet med de traditionelle monoaminerge og glutamaterge abnormiteter rapporteret i disse lidelser (figur 1 og 2). Den terapeutiske rolle af antiinflammatoriske midler i psykiatriske lidelser gennemgås også.
 Astroglial & Oligodendroglial Histopatologi
Astroglial & Oligodendroglial Histopatologi
Astroglia og oligodendroglia er afgørende for neurale metaboliske homeostase, adfærd og højere kognitive funktioner [54-56,133-136]. Normale hvilende astroglia giver energi og trofisk støtte til neuroner, regulerer synaptisk neurotransmission (figur 2), synaptogenese, cerebral blodgennemstrømning og opretholder BBB-integritet [134,136,137]. Modne oligodendroglia giver energi og trofisk støtte til neuroner og opretholder BBB-integritet og regulerer aksonal reparation�og myelinisering af hvide stof-kanaler, der giver inter- og intra-hemisfærisk forbindelse [54-56]. Både astroglia og oligodendroglia producerer antiinflammatoriske cytokiner, der kan nedregulere skadelig inflammation [52,55].
Ved MDD er astroglialt tab et konsekvent post-mortem fund i funktionelt relevante områder, herunder den forreste cingulate cortex, præfrontale cortex, amygdala og hvid substans [35-38,42-46,55,138-147], med få undtagelser [42,43]. Post-mortem undersøgelser afslørede reduceret glial fibrillært surt protein (GFAP)-positiv astroglial tæthed primært i den præfrontale cortex [37,38] og amygdala [36]. En stor proteomisk analyse af frontale cortex fra deprimerede patienter viste signifikante reduktioner i tre GFAP-isoformer [39]. Selvom der i en undersøgelse, der ikke rapporterede noget signifikant glialtab, afslørede undergruppeanalyse et signifikant fald (75%) i GFAP-positiv astroglial tæthed blandt forsøgspersoner yngre end 45 år [35]. En morfometrisk undersøgelse viste på samme måde ingen ændringer i glial tæthed i sen-life MDD hjerner [148]. Vi antager, at det tilsyneladende fravær af astroglialt tab blandt ældre MDD-patienter kan afspejle sekundær astrogliose [35], der er forbundet med ældre alder [42,50] snarere end en sand negativ.
Dyreforsøg er i overensstemmelse med humane undersøgelser, der viser astroglialt tab i MDD. Wistar-Kyoto-rotter, der er kendt for at udvise depressiv-lignende adfærd, afslørede reduceret astroglial tæthed i de samme områder som observeret hos mennesker [40]. Administration af det astroglialtoksiske middel, L-alfa-aminoadipinsyre, inducerer depressive-lignende symptomer hos rotter, hvilket tyder på, at astroglialt tab er patogent ved MDD [41].
Post-mortem undersøgelser af MDD-patienter dokumenterede reduceret oligodendroglial tæthed i den præfrontale cortex og amygdala [54-57,66], som kan korrelere med hjerne-MR-fokale hvide stof-ændringer, som lejlighedsvis observeres hos nogle MDD-patienter [57]. Mikrovaskulære abnormiteter kan dog også bidrage til disse ændringer [57].
I BPD viser nogle undersøgelser signifikant gliatab [138,143,149,150], mens andre ikke gør [37,44-46]. Disse inkonsekvente resultater kan skyldes manglende kontrol for: 1) behandling med stemningsstabilisatorer, fordi post-hoc-analyse rapporteret af nogle undersøgelser viste signifikant reduktion i gliatab først efter kontrol med behandling med lithium og valproinsyre [46]; 2) familiære former for BPD, da gliatab er særligt fremtrædende blandt BPD-patienter med en stærk familiehistorie [143]; og/eller 3) den dominerende tilstand af depression versus mani, da gliatab er hyppigt ved MDD [35-38,42-46,55,138-147]. Hvorvidt astroglia eller oligodendroglia tegner sig for størstedelen af gliatab er uklart; mens proteomisk analyse afslørede et signifikant fald i en astroglial GFAP isoform [39], fandt flere andre post-mortem undersøgelser enten uændret [36,37] eller reduceret GFAP-positiv astroglial ekspression i den orbitrofrontale cortex [47], eller reduceret oligodendroglial tæthed [54-56,58,59].
Ved skizofreni er astroglialt tab et inkonsekvent fund [48,150]. Mens nogle undersøgelser ikke viste noget signifikant astroglialt tab [42,50,51], fandt flere andre reduceret astroglial tæthed [37,38,43,44,48,49,151] og signifikante reduktioner i to GFAP isoformer [39]. Inkonsekvente fund kan skyldes: 1) MDD-komorbiditet, som ofte er forbundet med gliatab; 2) aldersvariation, da ældre patienter har øget GFAP-positiv astroglia [35,42,50]; 3) regional [150] og kortikal lagvariabilitet [48]; 4) behandling med antipsykotiske lægemidler, da eksperimentelle undersøgelser viser både reduceret [152] og øget [153] astroglial-densitet relateret til kronisk antipsykotisk behandling [70]; og 5) sygdomstilstand (for eksempel suicidal kontra ikke-suicidal adfærd) [154]. Post-mortem undersøgelser dokumenterede oligodendroglialt tab [54,56,60-65,148,155,156], især i den præfrontale cortex, anterior cingulate cortex og hippocampus [148]. Ultrastrukturel undersøgelse af den præfrontale region viste unormalt myelinerede fibre i både grå og hvid substans; både alder og varighed af sygdom var positivt korreleret med abnormiteterne i den hvide substans [157].
I modsætning til neurodegenerative lidelser, der almindeligvis er forbundet med astroglial spredning [136], er psykiatriske lidelser i stedet forbundet med enten reduceret eller uændret astroglial tæthed [138]. Manglen på øget glialtæthed i tidligt opståede psykiatriske lidelser [44,138] kan afspejle den langsommere hastighed af degenerativ progression i psykiatriske sygdomme [138].
Vi postulerer, at degenerative ændringer forbundet med psykiatriske lidelser er mere subtile og ikke alvorlige nok til at fremkalde astrogliale intracellulære transkriptionsfaktorer, som positivt regulerer astrogliose, herunder signaltransduceraktivator af transkription 3 og nuklear faktor kappa B (NF-?B) [136].
Mens størstedelen af post-mortem undersøgelser fokuserede på ændringen af glialtæthed i MDD, BPD og skizofreni, beskrev andre ændring af gliacellemorfologi med blandede fund. Ved MDD og BPD er glialstørrelsen enten øget eller uændret [55]. En undersøgelse fandt reduceret gliastørrelse ved BPD og skizofreni, men ikke ved MDD [43]. En post-mortem undersøgelse af deprimerede patienter, der begik selvmord, fandt øget astroglial størrelse i den forreste cingulate hvide substans, men ikke i cortex [158]. En undersøgelse af skizofrene forsøgspersoner fandt markant nedsat astroglial størrelse i lag V af den dorsolaterale præfrontale cortex, uanset at astroglial tæthed er dobbelt så stor som kontrollerne i det samme lag [48]. De blandede resultater kan delvist afspejle tidligere undersøgelser af gliale ændringer i psykiatriske sygdomme, der ikke specificerede astroglia versus oligodendroglia [148].
Gliatab ved psykiatriske sygdomme kan bidrage til neuroinflammation gennem flere mekanismer, herunder unormale cytokinniveauer (se afsnittet Cytokin), dysreguleret glutamatmetabolisme (se afsnittet Glutamat), forhøjet S100B-protein (se afsnittet S100B),�og ændret BBB-funktion (se afsnittet Blod-hjernebarriere), hvilket resulterer i svækket kognition og adfærd [44,45,54,133,159].
Mikroglial histopatologi
Microglia er de fastboende immunceller i CNS. De giver løbende immunovervågning og regulerer udviklingsmæssig synaptisk beskæring [160,161]. CNS-skade transformerer forgrenede hvilende mikroglia til aktiverede aflange stavformede og makrofaglignende fagocytiske amøboidceller, der prolifererer og migrerer mod skadestedet langs kemotaktiske gradienter (det vil sige mikroglial aktivering og proliferation (MAP)) [161]. Humane mikrogliaceller udtrykker NMDAR'er, der kan mediere MAP, hvilket fører til neuronal skade [162].
Ved MDD, BPD og skizofreni er resultaterne af post-mortem undersøgelser, der undersøger tilstedeværelsen af MAP, blandede. Post-mortem undersøgelser afslørede forhøjet MAP hos kun én ud af fem MDD-personer [67]. Hos nogle patienter med BPD-lidelse blev øget humant leukocytantigen-DR-positivt mikroglia, der viser fortykkede processer, dokumenteret i frontal cortex [69]. I skizofreni, mens nogle undersøgelser rapporterede forhøjet MAP i forhold til kontroller, viste andre ingen forskel mellem grupperne [22,67,70]. I en post-mortem undersøgelse, der vurderer MAP i MDD og BPD; quinolinsyre-positiv mikroglialcelletæthed blev øget i den subgenuelle anterior cingulate cortex og anterior midcingulate cortex hos MDD- og BPD-patienter, der begik selvmord i forhold til kontroller [53]. Post-hoc-analyse afslørede, at denne øgede MAP udelukkende kunne tilskrives MDD og ikke BPD, da den positive mikrogliale immunfarvning hos MDD-personer var signifikant større end den i BPD-undergruppen i både den subgenuelle anterior cingulate og midcingulate cortex, og da mikroglia-densiteten var ens i både BPD og kontrolgrupperne [53]. En undersøgelse, der sammenlignede alle tre lidelser (ni MDD, fem BPD, fjorten skizofreni, ti raske kontroller) viste ingen signifikant forskel i mikroglial tæthed på tværs af de fire grupper [68].
Disse blandede resultater kan tilskrives variable mikrogliale immunologiske markører anvendt blandt forskellige undersøgelser [70] og/eller manglende kontrol for sygdommens sværhedsgrad [22,53,68]. Navnlig dokumenterede tre post-mortem undersøgelser af MDD og skizofrene forsøgspersoner en stærk positiv sammenhæng mellem MAP og suicidalitet i den forreste cingulate cortex og mediodorsal thalamus, uafhængig af psykiatrisk diagnose [22,53,68]. MAP kan således være en tilstand snarere end en egenskabsmarkør for MDD og skizofreni.
I OCD tyder dyremodeller på, at dysfunktion og reduktion af visse mikrogliale fænotyper, såsom dem, der udtrykker Hoxb8-genet, som koder for homeobox-transkriptionsfaktor, kan forårsage OCD-lignende adfærd [71,72].
Hoxb8 knockout-mus udviser overdreven plejeadfærd og angst i forbindelse med reduceret mikroglial tæthed [71,72]. Denne overdrevne plejeadfærd ligner adfærdsegenskaberne ved menneskelig OCD. Hoxb8-injektion hos voksne Hoxb8-knockout-mus reverserer mikroglialt tab og genopretter normal adfærd [71,72]. Rollen af disse specifikke mikrogliale fænotyper i human OCD er uklar.
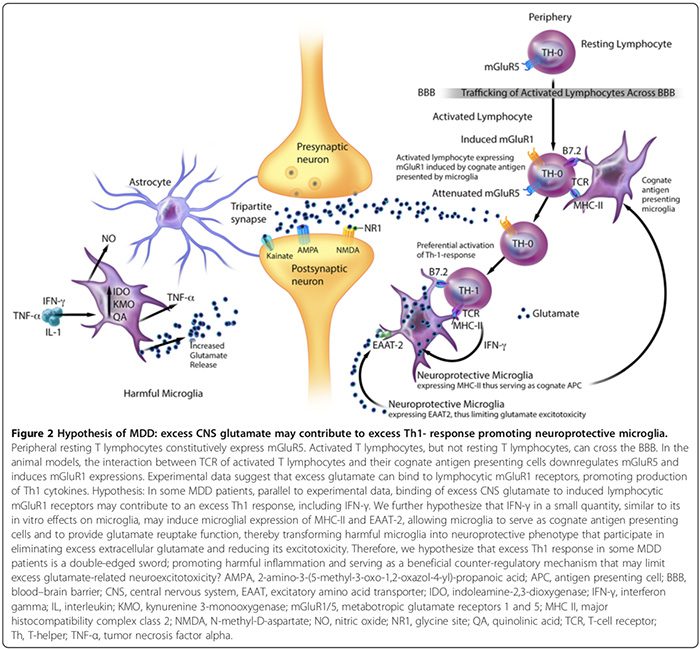
Eksperimentelle data tyder på, at MAP omfatter karakteristiske skadelige og neurobeskyttende fænotyper (figur 2). Skadelige mikroglia udtrykker ikke større histokompatibilitetskompleks II (MHC-II) og kan derfor ikke fungere som antigenpræsenterende celler (APC) [163,164]; de fremmer skadelige virkninger [17,69,165] gennem proinflammatorisk cytokinproduktion, nitrogenoxidsyntase-signalering [17,166], fremmer glial og BBB-pericyt/endotel cyclooxygenase-2 (COX-2) ekspression [167], inducerer astroglial S100B sekretion (se S100B sektion og mikro17,136,168,169B sekretion) 2]. Skadelige mikroglia udskiller også prostaglandin E-2 (PGE-2), der fremmer proinflammatorisk cytokinproduktion, hvilket igen øger PGE-29-niveauet i en feed-forward-cyklus [2]. Yderligere stimulerer PGE-2 COX-2-ekspression, som medierer omdannelsen af arachidonsyre til PGE-29, hvilket opsætter en anden feed-forward-cyklus [XNUMX].
Neuroprotektive mikroglia kan derimod: 1) udtrykke MHC-II in vivo og in vitro [163,166] og fungere som beslægtet APC (figur 2) [163,164,166]; 2) lette heling og begrænse neuronal skade ved at fremme sekretion af antiinflammatoriske cytokiner [17], hjerneafledt neurotrofisk faktor [17] og insulinlignende vækstfaktor-1 [166]; og 3) udtrykker excitatorisk aminosyretransporter-2 (EAAT2), der eliminerer overskydende ekstracellulær glutamat [163,166] og fremmer neurobeskyttende T-lymfocytisk autoimmunitet (figur 2) [163,164]. Der er imidlertid behov for flere undersøgelser for at bekræfte den medvirkende rolle af neurobeskyttende mikroglia til neuropsykiatriske lidelser hos mennesker.
In vitro dyreforsøg tyder på, at forholdet mellem skadelige versus neurobeskyttende mikroglia kan påvirkes af nettoeffekten af inflammatoriske modreguleringsmekanismer [15,74,164,166]. Disse mekanismer omfatter antallet af neurobeskyttende CD4+CD25+FOXP3+ T-regulatoriske celler ((T regs) figur 1) [15,74,164,166] og hjernecytokinniveauer; lav IFN-? niveauer kan fremme neurobeskyttende mikroglia (figur 2) [166], hvorimod høje niveauer kan fremme den skadelige fænotype [166].
Cytokinernes rolle
Proinflammatoriske cytokiner inkluderer IL-1a, IL-2, IL-6, TNF-a og IFN-?. De udskilles primært af mikroglia, Th1-lymfocytter og M1-fænotypemonocytter/makrofager (figur 1) [15,170]. De fremmer skadelig betændelse. Antiinflammatoriske cytokiner omfatter IL-4, IL-5 og IL-10. De udskilles primært af astroglia,�Th2 lymfocytter, T regs og M2 fænotype monocytter/makrofager [15,52,74]. De kan begrænse skadelig inflammation [15,74] ved at omdanne den proinflammatoriske M1-fænotype til den gavnlige antiinflammatoriske M2-fænotype [15], og potentielt ved at fremme den neurobeskyttende mikrogliale fænotype [15,17,74,163,166]. Rollen af proinflammatoriske/antiinflammatoriske cytokiner i psykiatriske lidelser understøttes af flere bevislinjer (Figur 1, Tabel 2) [15,17,29,52,74].
I MDD bekræftede den seneste meta-analyse (29 undersøgelser, 822 MDD, 726 sunde kontroller) af serum proinflammatoriske cytokiner, at opløselig IL-2-receptor, IL-6 og TNF-? niveauer er øget i MDD (egenskabsmarkører) [91], mens IL-1?, IL-2, IL-4, IL-8 og IL-10 ikke er statistisk forskellige fra kontroller [91]. I et primært cytokinstudie, der sammenlignede MDD-undergrupper (47 suicidal-MDD, 17 ikke-suicidal-MDD, 16 sundhedskontroller), både sera IL-6 og TNF-? var signifikant højere, mens IL-2-niveauer var signifikant lavere hos MDD-personer, der begik selvmord i forhold til begge andre grupper [96]. Dette fund tyder på, at IL-6 og TNF-? er også tilstandsmarkører for MDD [96]. Faldet i serum-IL-2-niveauer forbundet med akut selvmordsadfærd kan afspejle øget binding til dens opregulerede receptor i hjernen; parallelt med den førnævnte meta-analyse, der viser øget opløselig IL-2-receptor i MDD [91]. Undersøgelser, der undersøgte den kliniske betydning af cytokiner i MDD viste, at serumcytokinniveauer er forhøjede under akutte depressive episoder [171,172] og normaliseres efter vellykket, men ikke fejlslagen, behandling med antidepressiva [17] og elektrokonvulsiv terapi [29]; disse fund tyder på en mulig patogen rolle for cytokiner.
I BPD blev serumcytokinændringer opsummeret i en nylig gennemgang; TNF-a, IL-6 og IL-8 er forhøjet under maniske og depressive faser, hvorimod IL-2, IL-4 og IL-6 er forhøjet under mani [92]. Andre undersøgelser viste, at sera IL-1? og IL-1-receptorniveauer er ikke statistisk forskellige fra raske kontroller [92], selvom vævsundersøgelser dokumenterede øgede niveauer af IL-1? og IL-1 receptor i BPD frontal cortex [69].
Ved skizofreni er resultaterne fra undersøgelser, der undersøger cytokinabnormiteter, modstridende (tabel 2). Mens nogle undersøgelser fandt både nedsat serum proinflammatorisk (IL-2, IFN-?) og øget serum og CSF antiinflammatoriske cytokiner (IL-10) [52], fandt andre forhøjede serum pro- og antiinflammatoriske cytokiner med en proinflammatorisk type dominans [22,173,174]. Én cytokin-meta-analyse (62 undersøgelser, 2,298 skizofreni, 858 raske kontroller) viste øgede niveauer af IL-1R-antagonist, sIL-2R og IL-6 [174]. Denne undersøgelse redegjorde dog ikke for brugen af antipsykotika, som menes at øge proinflammatorisk cytokinproduktion [52]. En nyere cytokin-meta-analyse (40 undersøgelser, 2,572 skizofrene,�4,401 kontroller), der tegnede sig for antipsykotika, fandt, at TNF-a, IFN-a, IL-12 og sIL-2R er konsekvent forhøjede ved kronisk skizofreni uafhængigt af sygdomsaktivitet (egenskabsmarkører), mens IL-1?, IL-6 og transformerende vækstfaktor beta korrelerer positivt med sygdomsaktivitet] [173-markører. Cellekulturer af mononukleære celler fra perifert blod (PBMC) opnået fra skizofrene patienter producerede højere niveauer af IL-8 og IL-1? spontant såvel som efter stimulering af LPS, hvilket tyder på en rolle for aktiverede monocytter/makrofager i skizofrenipatologi [175].
Ved OCD er resultaterne fra en tilfældig undersøgelse af sera- og CSF-cytokiner og LPS-stimulerede PBMC-undersøgelser inkonsistente [93-95,176-179]. Der er en sammenhæng mellem OCD og en funktionel polymorfi i promotorregionen af TNF-? gen [34], selv om lavenergiundersøgelser ikke bekræftede denne sammenhæng [180]. Derfor er de blandede resultater fra undersøgelser, der dokumenterer enten øget eller nedsat TNF-? cytokinniveauer [93,176-178] kan afspejle deres variable inklusion af undergruppen af OCD-personer med denne særlige polymorfi i deres kohorter.
Cytokinresponspolarisering ved svær depression og skizofreni
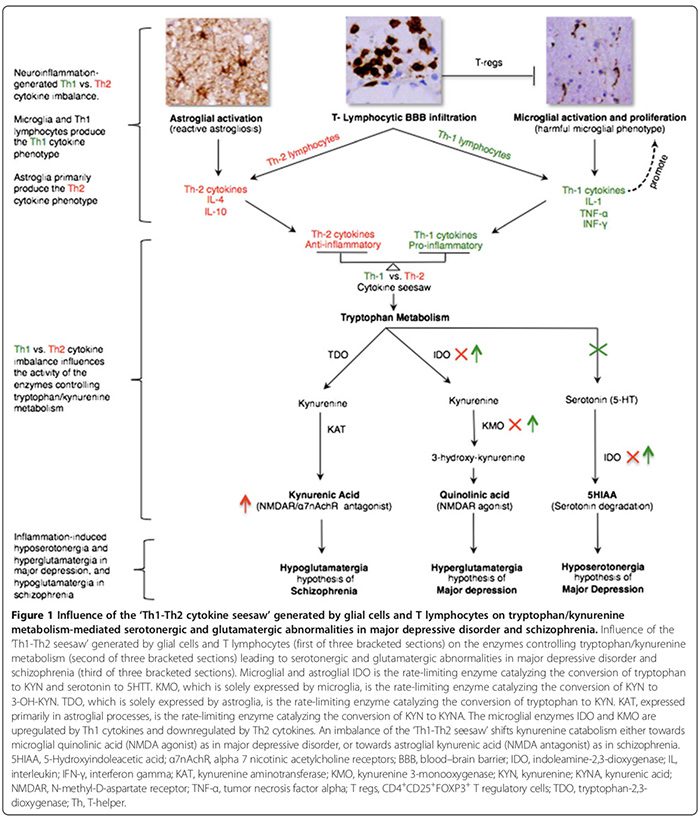
Cytokinresponsfænotyper klassificeres som enten proinflammatorisk Th1 (IL-2, IFN-?) eller antiinflammatorisk Th2 (IL-4, IL-5, IL-10) i henhold til de immunfunktioner, de regulerer. Mens Th1-cytokiner regulerer cellemedieret immunitet rettet mod intracellulære antigener, regulerer Th2-cytokiner humoral immunitet rettet mod ekstracellulære antigener [29,52]. Th1-cytokiner produceres af Th1-lymfocytter og M1-monocytter, mens Th2-cytokiner produceres af Th2-lymfocytter og M2-monocytter [29,52]. I hjernen udskiller mikroglia overvejende Th1-cytokiner, hvorimod astroglia overvejende udskiller Th2-cytokiner [29,52]. Det gensidige forhold mellem Th1:Th2-cytokiner, herefter �Th1-Th2-vippe,� er påvirket af andelen af aktiverede mikroglia (overskydende Th1) til astroglia (overskydende Th2) og samspillet mellem aktiverede T-celler og for høje CNS-glutamatniveauer, som vi antog for at favorisere Th1-respons (Figure2)
Th1-Th2 vippeubalancen kan påvirke tryptofan metabolisme ved at ændre dets enzymer [21,52] og derved flytte tryptophan katabolisme mod kynurenin (KYN) og KYN katabolisme mod en af dets to nedstrøms metabolitter; microglia quinolinsyre, der er Th1-responsmedieret eller astroglial kynureninsyre (KYNA) (figur 1), der er Th2-responsmedieret [21,29,170].
Tryptophan metabolisme enzymer påvirket af Th1-Th2 vippe omfatter (figur 1): indolamin 2,3-dioxygenase (IDO) udtrykt af mikroglia og astroglia, de hastighedsbegrænsende enzymer, der medierer omdannelsen af tryptophan til KYN og serotonin til 5-hydroxyindoleddikesyre�[21,29]. Kynurenin 3-monooxygenase (KMO), udelukkende udtrykt af mikroglia, er det hastighedsbegrænsende enzym, der omdanner KYN til 3-hydroxykynurenin (3-OH-KYN), som metaboliseres yderligere til quinolinsyre [21,29]. Tryptophan-2,3-dioxygenase (TDO), udelukkende udtrykt af astroglia, er det hastighedsbegrænsende enzym, der omdanner�tryptofan til KYN [21,29]. Kynureninaminotransferase (KAT), primært udtrykt i astrogliale processer, er det hastighedsbegrænsende enzym, der medierer omdannelsen af KYN til KYNA [21,29].
Th1 cytokiner aktiverer mikroglial IDO og KMO, og flytter mikroglial KYN katabolisme mod quinolinisk�syre (NMDAR agonist) syntese, mens Th2 cytokiner inaktiverer mikroglial IDO og KMO, hvilket skifter astroglial KYN katabolisme mod TDO- og KAT-medieret KYNA (NMDAR antagonist) syntese (Figur 1) [21,29].
Th1- og Th2-dominerende immunfænotyper er blevet foreslået for henholdsvis MDD og skizofreni baseret på perifere, snarere end CNS, cytokinmønstre [52,173]. Vi mener, at perifere cytokinmønstre er upålidelige surrogatmarkører for dem i CNS. Faktisk kan perifere cytokinniveauer påvirkes af mange ekstra-CNS-variabler, som ikke konsekvent kontrolleres for i flere af de perifere cytokinundersøgelser, herunder: 1) alder, kropsmasseindeks, psykotrope medicin, rygning, stress og døgnudsving; 2) indflydelse af�sygdomsaktivitet/tilstand på produktionen af udvalgte cytokinsyntese [95,173]; og 3) virkningerne af psykotrope midler på cytokinproduktion [52]. De korte halveringstider og den hurtige omsætning af serumcytokiner [181] (for eksempel 18 minutter for TNF-a [182] versus 60 minutter for IL-10 [183]), kan yderligere begrænse pålideligheden af at fortolke deres niveauer målt fra tilfældig seraprøvetagning.
Ved MDD er der konsensus om, at en proinflammatorisk Th1-immunfænotype-respons dominerer (tabel 2) [17,29]. Høje niveauer af quinolinsyre i post-mortem MDD hjerner [53], tyder på tilstedeværelsen af en opreguleret Th1 respons (figur 1) [21,29]. Forhøjet CNS quinolinsyre kan fremme calciumtilstrømningsmedieret apoptose af human astroglia [184], som hypotetisk kan sløve �astroglia-afledt Th2-respons [29], vælter Th1 versus Th2 vippebalance til fordel for den mikrogliale Th1-respons. CNS-hyposerotonergi [29] tilføjer yderligere støtte til et overskydende Th1-respons, som er vist at reducere CNS-serotoninsyntese [185] og øge dets nedbrydning (figur 1) [21,29].
CNS-hyperglutamatergi kan også bidrage til et overskydende Th1-respons i hjernen (figur 2). En in vitro undersøgelse tyder på, at de perifere hvilende T-lymfocytter konstitutivt udtrykker metabotropisk glutamatreceptor 5 (mGluR5) [164], hvis binding til glutamat hæmmer lymfocytisk IL-6-frigivelse og derved nedregulerer autoreaktiv T-effektorcelleproliferation [164]. Aktiverede T-lymfocytter, men ikke hvilende T-lymfocytter, kan krydse BBB [37].
Eksperimentelle data tyder på, at interaktionen mellem T-cellereceptorer af aktiverede T-lymfocytter og deres beslægtede antigenpræsenterende celler kan nedregulere mGluR5 og inducere mGluR1-ekspressioner [164]. I dyremodeller fremmer binding af overskydende glutamat til lymfocytiske mGluR1-receptorer produktionen af Th1-cytokiner, herunder IFN-? [164].
Vi antager, at hos nogle MDD-patienter, parallelt med eksperimentelle data [164], kan bindingen af overskydende CNS-glutamat til inducerede lymfocytiske mGluR1-receptorer bidrage til et overskydende Th1-respons, herunder IFN-? (Figur 2). Vi spekulerer i, at IFN-? i en lille mængde, svarende til dens in vitro-effekter på mikroglia [166], kan inducere mikroglial ekspression af MHC-II og EAAT2 [163,166], hvilket gør det muligt for mikroglia at tjene som beslægtede antigenpræsenterende celler og give glutamatgenoptagelsesfunktion [163,164,166], derved transformerer 163,166, neuroprotetive mikro163,164,166, harmisk [1] som deltager i eliminering af overskydende ekstracellulært glutamat [2]. Derfor antager vi også, at overskydende ThXNUMX-respons i undergrupper af MDD-patienter er et tveægget sværd, der fremmer skadelig inflammation og tjener som en gavnlig modreguleringsmekanisme, der kan begrænse overskydende glutamatrelateret neuroexcitotoksicitet (figur XNUMX).
I skizofreni, mens nogle perifere cytokinundersøgelser tyder på overvægten af en antiinflammatorisk Th2 immunfænotype/respons [52], modbeviser andre dette [173,174]. Men vi er enige med forfatterne, der antog, at Th2-responset er den dominerende fænotype i skizofreni [52]. Forhøjede hjerne-, CSF- og serumniveauer af KYNA [21,52] tyder på nedregulering af mikroglial IDO og KMO, som er en funktion af Th2-respons, der skifter astroglial KYN-katabolisme mod KYNA-syntese (figur 1) [21,52]. Reduceret KMO-aktivitet og KMO-mRNA-ekspression i post-mortem skizofrene hjerner [73] er i overensstemmelse med overskydende Th2-respons (figur 1). Øget prævalens af Th2-medierede humorale immunitetsabnormiteter i undergrupper af skizofrenipatienter�som påvist af øget B-celletal [21,76], øget�produktion af autoantistoffer inklusive antivirale antistoffer [76] og øget immunglobulin E [52] tilføjer yderligere støtte til Th2-responsdominanshypotesen.
Neuroinflammation og CNS Glutamat Dysregulering
Glutamat medierer kognition og adfærd [186]. Synaptiske glutamatniveauer reguleres af natriumafhængige gliale og neuronale EAAT'er med høj affinitet, nemlig XAG-systemet, der er ansvarligt for glutamatgenoptagelse/aspartatfrigivelse [137,164] og det natrium-uafhængige astrogliale glutamat/cystin antiporter system (Xc-) ansvarlig for [164 genoptagelse]/cystine1 reuptake. Astroglial EAAT2 og EAAT90 giver mere end 79% af glutamatgenoptagelsen [XNUMX].
Neuroinflammation kan ændre glutamatmetabolismen og funktionen af dets transportører [15,29,187,188], hvilket producerer kognitive, adfærdsmæssige og psykiatriske svækkelser [15,21,29,79,186,188,189]. Abnormiteter af EAATs funktion/udtryk og glutamatmetabolisme i MDD, BPD, skizofreni og OCD er opsummeret i tabel 2.
Ved MDD er der evidens for kortikal hyperglutamatergi (tabel 2). Kortikale glutamatniveauer korrelerede positivt med sværhedsgraden af depressive symptomer, og en fem-ugers behandling med antidepressiva reducerede serum glutamatkoncentrationer [85,86]. En enkelt dosis ketamin, en potent NMDAR-antagonist, kan vende refraktær MDD i en uge [17,21,29,85]. Overskydende CNS-glutamatniveauer kan inducere neurotoksicitetsmedieret inflammation [163,164,188], herunder et proinflammatorisk Th1-respons (figur 2) [164].
Begrænset in vitro-evidens tyder på, at inflammation/proinflammatoriske cytokiner kan øge CNS-glutamatniveauer [188] i en feed-forward-cyklus gennem flere potentielle mekanismer: 1) proinflammatoriske cytokiner kan hæmme [15,17,168] og reversere [45,137] astroglial EAAT-medieret glutamatfunktion; 2) proinflammatoriske cytokiner kan forbedre mikroglial quinolinsyresyntese [53], som eksperimentelt er blevet vist at fremme synaptosomal glutamatfrigivelse [15,17,29,190]; 3) øget COX-2/PGE-2 og TNF-? niveauer kan inducere calciumtilstrømning [137], hvilket, baseret på in vitro data, kan øge astroglial glutamat og D-serin frigivelse [191]; og 4) aktiverede mikroglia kan udtrykke overskydende Xc-antiporter-systemer, der medierer glutamatfrigivelse [164,192].
Ved skizofreni findes præfrontal kortikal hypoglutamatergi [87,90,193,194] (tabel 2) og reduceret NMDAR-funktionalitet [5]. Nyere H1 magnetisk resonansspektroskopi (MRS) meta-analyse (28 undersøgelser, 647 skizofreni, 608 kontrol) bekræftede nedsat glutamat og øgede glutaminniveauer i den mediale frontale cortex [90]. Den medvirkende rolle af inflammation til hypoglutamatergi er ikke bevist. Forhøjet KYNA-syntese i skizofreni-hjerner [21,52], typisk en funktion af Th2-respons (figur 1), kan hæmme NR1-underenhed af NMDAR og alfa 7 nikotinisk�acetylcholinreceptor (?7nAchR) [195], hvilket fører til nedsat NMDAR-funktion og reduceret a7nAchR-medieret glutamatfrigivelse [195].
Ved BPD og OCD tyder data på CNS-kortikal hyperglutamatergi i begge lidelser (tabel 2) [78,84,88,131]. Bidraget fra inflammation (BPD og OCD) og autoantistoffer (OCD) [7,77,84,88,130] til øgede CNS-glutamatniveauer kræver yderligere undersøgelse.
S100B's rolle
S100B er et 10 kDa calciumbindende protein produceret af astroglia, oligodendroglia og choroid plexus ependymale celler [196]. Det medierer dets virkninger på de omgivende neuroner og glia via receptoren for avanceret glycation slutprodukt [196]. Nanomolære ekstracellulære S100B-niveauer giver gavnlige neurotrofiske virkninger, begrænser stressrelateret neuronal skade, hæmmer mikroglial TNF-? frigivelse og øge astroglial glutamatgenoptagelse [196]. Mikromolære S100B-koncentrationer, overvejende produceret af aktiverede astroglia og lymfocytter [196,197], har skadelige virkninger transduceret af receptoren for avanceret glycation slutprodukt, der omfatter neuronal apoptose, produktion af COX-2/PGE-2, IL-1? og inducerbare nitrogenoxidarter og opregulering af monocytisk/mikroglial TNF-? sekretion [21,196,198].
Serum og især CSF og hjernevæv S100B niveauer er indikatorer for glial (overvejende astroglial) aktivering [199]. Ved MDD og psykose korrelerer serum S100B-niveauer positivt med sværhedsgraden af suicidalitet, uafhængigt af psykiatrisk diagnose [200]. Post-mortem analyse af S100B viste nedsatte niveauer i den dorsolaterale præfrontale cortex af MDD og BPD og øgede niveauer i parietal cortex af BPD [196].
Meta-analyse (193 stemningslidelser, 132 raske kontroller) bekræftede forhøjede serum- og CSF S100B-niveauer i stemningslidelser, især under akutte depressive episoder og mani [201].
Ved skizofreni er hjerne, CSF og serum S100B niveauer forhøjede [199,202]. Meta-analyse (12 undersøgelser, 380 skizofreni, 358 raske kontroller) bekræftede forhøjede serum-S100B-niveauer ved skizofreni [203]. I post-mortem hjerner hos skizofrenipatienter findes S100B-immunoreaktive astroglia i områder, der er impliceret i skizofreni, herunder forreste cingulate cortex, dorsolateral præfrontal cortex, orbitofrontal cortex og hippocampi [154]. Forhøjede S100B-niveauer korrelerer med paranoid [154] og negativistisk psykose [204], nedsat kognition, dårlig terapeutisk respons og varighed af sygdom [202]. Genetiske polymorfismer i S100B [32] og receptor for avancerede glycation-slutproduktgener i skizofreni-kohorter (tabel 2) [32,33,205] tyder på, at disse abnormiteter sandsynligvis er primære/patogene snarere end sekundære/biomarkører. Faktisk tyder faldet i serum S100B-niveauer efter behandling med antidepressiva [201] og antipsykotika [196]�en vis klinisk relevans af S100B for patofysiologien af psykiatriske lidelser.
Neuroinflammation og øget oxidativ stress
Oxidativt stress er en tilstand, hvor et overskud af oxidanter beskadiger eller modificerer biologiske makromolekyler såsom lipider, proteiner og DNA [206-209]. Dette overskud skyldes øget oxidantproduktion, nedsat oxidanteliminering, defekt antioxidantforsvar eller en kombination deraf [206-209]. Hjernen er særligt sårbar over for oxidativt stress på grund af: 1) forhøjede mængder af peroxiderbare flerumættede fedtsyrer; 2) relativt højt indhold af spormineraler, der inducerer lipidperoxidation og oxygenradikaler (f.eks. jern, kobber); 3) høj iltudnyttelse; og 3) begrænsede antioxidationsmekanismer [206,207].
Overdreven oxidativ stress kan forekomme ved MDD [206], BPD [206,207], skizofreni [207,209] og OCD [206,208]. Perifere markører for oxidative forstyrrelser omfatter øgede lipidperoxidationsprodukter (for eksempel malondialdehyd og 4-hydroxy-2-nonenal), øgede nitrogenoxid (NO) metabolitter, nedsatte antioxidanter (for eksempel glutathion) og ændrede antioxidantenzymniveauer [206,207].
I MDD korrelerer øget superoxidradikalanionproduktion med øget oxidationsmedieret neutrofil apoptose [206]. Serumniveauer af antioxidantenzymer (f.eks. superoxiddismutase-1) er forhøjede under akutte depressive episoder og normaliseres efter behandling med selektiv serotonin-genoptagelseshæmmere (SSRI'er) [206]. Dette tyder på, at ved MDD er antioxidantenzymniveauer i serum en tilstandsmarkør, som kan afspejle en kompenserende mekanisme, der modvirker akutte stigninger i oxidativt stress. [206]. I skizofreni derimod er CSF-opløselige superoxiddismutase-1-niveauer væsentligt reduceret hos tidligt debuterende skizofrene patienter i forhold til kroniske skizofrene patienter og raske kontroller. Dette tyder på, at reducerede hjerneantioxidantenzymniveauer kan bidrage til oxidativ skade ved akut skizofreni [210], selvom større undersøgelser er nødvendige for at bekræfte dette fund.
Adskillige yderligere eksperimentelle og humane undersøgelser undersøgte mere detaljeret de mekanismer, der ligger til grund for patofysiologien af øget oxidativt stress i psykiatriske lidelser [206-262]. I dyremodeller af depression reduceres hjerneniveauer af glutathion, mens lipidperoxidation og NO-niveauer øges [206,262].
Postmortem undersøgelser viser reducerede hjerneniveauer af total glutathion hos MDD, BPD [206] og skizofrene forsøgspersoner [206,207]. Fibroblaster dyrket fra MDD-patienter viser øget oxidativt stress uafhængigt af glutathionniveauer [262], hvilket argumenterer imod en primær rolle af glutathiondepletering som den vigtigste mekanisme for oxidativt stress i depression.
Mikroglial aktivering kan øge oxidativt stress gennem sin produktion af proinflammatoriske cytokiner og NO [206-209]. Proinflammatoriske cytokiner og høje NO-niveauer kan fremme dannelsen af reaktive oxygenarter (ROS), som igen accelererer lipidperoxidation, beskadiger membranfosfolipider og deres membranbundne monoamin-neurotransmitterreceptorer og udtømmer endogene antioxidanter. Forøgede ROS-produkter kan øge mikroglial aktivering og øge proinflammatorisk produktion via stimulerende NF-?B [208], hvilket igen forstærker oxidativ skade [208], hvilket skaber potentialet for en patologisk positiv feedback-loop i nogle psykiatriske lidelser [206-209]. Selvom neuroinflammation kan øge hjernens glutamatniveauer [85,86], forbliver rollen af glutamaterg hyperaktivitet som årsag til oxidativt stress ubegrundet [207].
Mitokondriel dysfunktion kan bidrage til øget oxidativt stress i MDD, BPD og skizofreni [206]. Postmortem undersøgelser af disse lidelser afslører abnormiteter i mitokondrielt DNA, i overensstemmelse med den høje forekomst af psykiatriske forstyrrelser i primære mitokondrielle lidelser [206]. In vitro dyreforsøg viser, at proinflammatoriske cytokiner, såsom TNF-?, kan reducere mitokondriel tæthed og forringe mitokondriel oxidativ metabolisme [211,212], hvilket fører til øget ROS-produktion [206,213]. Disse eksperimentelle resultater kan antyde mekanistiske forbindelser mellem neuroinflammation, mitokondriel dysfunktion og oxidativ stress [206,213], hvilket fortjener yderligere undersøgelse af disse krydsende patogene veje i menneskelige psykiatriske lidelser.
Det neurale vævs sårbarhed over for oxidativ skade varierer mellem forskellige psykiatriske lidelser baseret på de neuroanatomiske, neurokemiske og molekylære veje involveret i den specifikke lidelse [207]. Behandlingseffekter kan også være kritiske, da foreløbige beviser tyder på, at antipsykotika, SSRI'er og humørstabilisatorer har antioxidantegenskaber [206,207,262]. Den terapeutiske rolle af adjuverende antioxidanter (f.eks. vitamin C og E) i psykiatriske lidelser mangler stadig at blive underbygget af kraftige randomiserede kliniske forsøg. N-acetylcystein viser de mest lovende resultater til dato, med flere randomiserede placebokontrollerede forsøg, der viser dets effektivitet ved MDD, BPD og skizofreni [207].
Dysfunktion af blod�hjernebarriere
BBB sikrer hjernens immunprivilegerede status ved at begrænse adgangen til perifere inflammatoriske mediatorer, herunder cytokiner og antistoffer, der kan svække neurotransmission [214,215]. Hypotesen om BBB-nedbrydning og dens rolle hos nogle psykiatriske patienter [60,214,216,217] er i overensstemmelse med den øgede forekomst af psykiatrisk komorbiditet i sygdomme forbundet med dets dysfunktion, herunder SLE [97], slagtilfælde [11],�epilepsi [218] og autoimmune encephalitider (tabel 1). Et forhøjet �CSF:serumalbumin ratio� hos patienter med MDD og skizofreni tyder på øget BBB-permeabilitet [214].
I en undersøgelse (63 psykiatriske forsøgspersoner, 4,100 kontroller) blev CSF-abnormaliteter, der tyder på BBB-beskadigelse, påvist hos 41 % af psykiatriske forsøgspersoner (14 MDD og BPD, 14 skizofreni), inklusive intratekal syntese af IgG, IgM og/eller IgA, mild CSF-celle tilstedeværelse af IgG til 5-8 mm celler ligoklonale bånd [3]. En post-mortem ultrastrukturel undersøgelse i skizofreni afslørede BBB ultrastrukturelle abnormiteter i de præfrontale og visuelle cortex, som omfattede vakuolær degeneration af endotelceller, astroglial-ende-fod-processer og fortykkelse og uregelmæssighed af den basale lamina [216]. Men i denne undersøgelse kommenterede forfatterne ikke på det potentielle bidrag af postmortem-ændringer til deres resultater. En anden undersøgelse, der undersøgte transkriptomi af BBB-endotelceller i skizofrene hjerner, identificerede signifikante forskelle mellem gener, der påvirker immunologisk funktion, som ikke blev påvist i kontroller [60].
Oxidationsmedieret endoteldysfunktion kan bidrage til patofysiologien af BBB-dysfunktion ved psykiatriske lidelser. Indirekte beviser fra kliniske og eksperimentelle undersøgelser i depression [219] og i mindre grad i skizofreni [220] tyder på, at øget oxidation kan bidrage til endotel dysfunktion. Endoteldysfunktion kan repræsentere en fælles mekanisme, der tegner sig for den kendte sammenhæng mellem depression og kardiovaskulær sygdom [219,221], som kan være relateret til nedsatte niveauer af vasodilator NO [221-223]. Eksperimentelle undersøgelser tyder på, at reducerede endotheliale NO-niveauer er mekanisk forbundet med afkoblingen af endotelnitrogenoxidsyntase (eNOS) fra dets essentielle co-faktor tetrahydrobiopterin (BH4), hvilket skifter dets substrat fra L-arginin til oxygen [224-226]. Ukoblet eNOS fremmer syntese af ROS (for eksempel superoxid) og reaktive nitrogenarter (RNS) (for eksempel peroxynitrit; et produkt af interaktionen af superoxid med NO) [227] snarere end NO, hvilket fører til oxidationsmedieret endoteldysfunktion [224-226].
Dyredata viste, at SSRI'er kunne genoprette mangelfulde endotheliale NO-niveauer [219], hvilket tyder på, at antioxidative mekanismer kan bidrage til deres antidepressive virkninger. Hos mennesker kan L-methylfolat forstærke anti-depressive virkninger af SSRI'er [228], formodentlig ved at øge niveauet af BH4, som er en essentiel cofaktor for eNOS genkoblingsmedieret antioxidation [229], såvel som for de hastighedsbegrænsende enzymer af monoamin (det vil sige, serotonin) synthese, [228aminepinephrine] [XNUMXaminpinephrine].
Tilsammen, både det nylige arbejde, der understreger rollen af ukoblet eNOS-induceret oxidativt stress i patogenesen af vaskulære sygdomme [230,231] ogepidemiologiske undersøgelser, der etablerer depression som en uafhængig risikofaktor for vaskulære patologier, såsom slagtilfælde og hjertesygdomme [219,221], tilføjer yderligere støtte til den kliniske relevans af ukoblet eNOS-medieret endotel oxidativ skade i depression. På trods af rigelig evidens for cytokinabnormaliteter i humane psykiatriske sygdomme og de eksperimentelle data, der viser, at proinflammatoriske cytokiner kan reducere eNOS-ekspression [212] og øge BBB-permeabilitet [215], mangler der humane beviser, der direkte forbinder overskydende proinflammatoriske cytokiner til eNOS-dysfunktion og/eller BBB-svækkelse.
Billeddannelse og behandling af betændelse ved psykiatrisk sygdom
Billeddannelse af neuroinflammation in situ
Klinisk kan neuroinflammationsbilleddannelse vise sig at være afgørende for at identificere den undergruppe af psykiatriske patienter med neuroinflammation, som med størst sandsynlighed vil reagere positivt på immunmodulerende terapier. Derudover kan sådan billeddannelse give klinikere mulighed for at overvåge neuroinflammationsrelateret sygdomsaktivitet og dens respons på immunterapi hos psykiatriske patienter. Billeddannende inflammation i den menneskelige hjerne har traditionelt været afhængig af MR- eller CT-visualisering af ekstravagerede intravenøse kontrastmidler, hvilket indikerer lokaliseret nedbrydning af BBB. Gadolinium-forstærket MR viser lejlighedsvis sådan nedbrydning i de limbiske regioner forbundet med følelsesmæssig behandling hos patienter med psykiatriske lidelser, der kan tilskrives paraneoplastiske eller andre encephalitider [107,109,113]. Så vidt vi ved, er unormal forbedring dog aldrig blevet påvist i nogen klassisk psykiatrisk lidelse [21,214,232], på trods af funktionelle [214,216] og ultrastrukturelle BBB-abnormiteter [60].
Hvorvidt subtile neuroinflammation kan visualiseres in vivo i klassiske psykiatriske lidelser er stadig ukendt. En lovende teknik er positronemissionstomografi (PET) ved hjælp af radiotracere, såsom C11-PK11195, som binder til translokatorproteinet, tidligere kendt som den perifere benzodiazepinreceptor, udtrykt af aktiverede mikroglia [233,234].
Ved at bruge denne metode blev patienter med skizofreni vist at have større mikroglial aktivering i hele cortex [235] og i hippocampus under akut psykose [236]. Et studie (14 skizofreni, 14 kontroller) fandt ingen signifikant forskel mellem [11C] DAA1106-binding ved skizofreni versus kontroller, men en direkte sammenhæng mellem [11C] DAA1106-binding og sværhedsgraden af positive symptomer og sygdomsvarighed ved skizofreni [236].
Efterforskere fra vores institution brugte C11-PK11195 PET til at demonstrere bi-hippocampus inflammation hos en patient med neuropsykiatrisk dysfunktion, herunder psykotisk MDD, epilepsi og anterograd amnesi, forbundet med anti-GAD antistoffer [237]. Dog har PK11195 PET�lave signal-til-støj egenskaber og kræver en on-site cyklotron.
Derfor afsættes forskning til at udvikle forbedrede translokatorproteinligander til PET og SPECT. Fremtidige kraftfulde post-mortem hjernevævsundersøgelser, der anvender proteinkvantificering, der sigter mod at belyse metaboliske og inflammatoriske veje, CNS-cytokiner og deres bindingsreceptorer i psykiatriske lidelser er nødvendige for at fremme vores forståelse af den autoimmune patofysiologi.
Rolle af antiinflammatoriske lægemidler i psykiatriske lidelser
Adskillige undersøgelser af mennesker og dyr tyder på, at visse antiinflammatoriske lægemidler kan spille en vigtig supplerende rolle i behandlingen af psykiatriske lidelser (tabel 3). Almindelige lægemidler er cyclooxygenasehæmmere (tabel 3) [238-245], minocyclin (tabel 3) [240-245], omega-3 fedtsyrer [246,247] og neurosteroider [248].
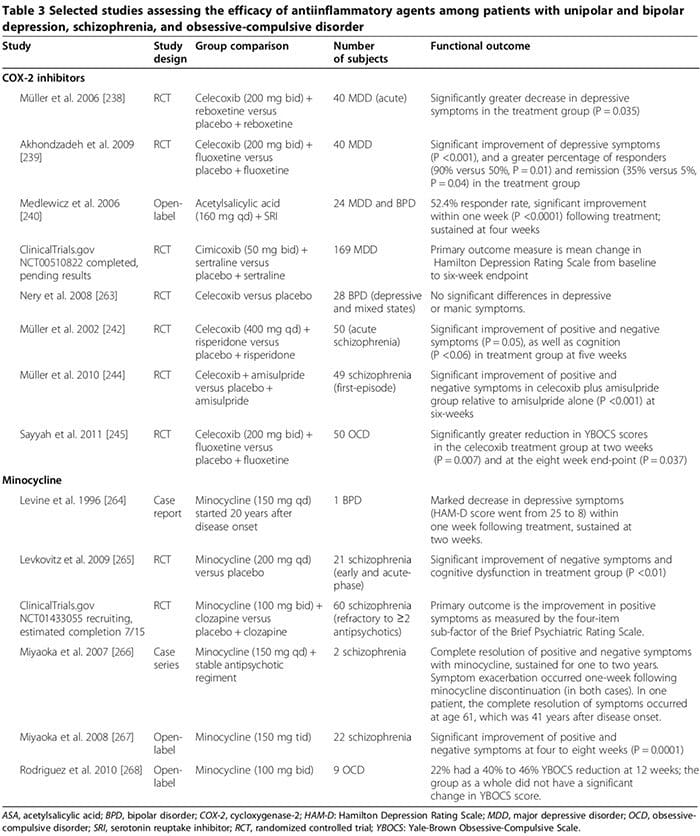 Adskillige humane undersøgelser viste, at COX-2-hæmmere kunne lindre psykiatriske symptomer på MDD, BPD, skizofreni og OCD (tabel 3) [248]. I modsætning hertil kan supplerende behandling med ikke-selektive COX-hæmmere (det vil sige non-steroide antiinflammatoriske lægemidler (NSAID'er)) reducere effektiviteten af SSRI'er [249,250]; to store forsøg rapporterede, at eksponering for NSAID'er (men ikke for hverken selektive COX-2-hæmmere eller salicylater) var forbundet med en signifikant forværring af depression blandt en undergruppe af studiedeltagere [249,250].
Adskillige humane undersøgelser viste, at COX-2-hæmmere kunne lindre psykiatriske symptomer på MDD, BPD, skizofreni og OCD (tabel 3) [248]. I modsætning hertil kan supplerende behandling med ikke-selektive COX-hæmmere (det vil sige non-steroide antiinflammatoriske lægemidler (NSAID'er)) reducere effektiviteten af SSRI'er [249,250]; to store forsøg rapporterede, at eksponering for NSAID'er (men ikke for hverken selektive COX-2-hæmmere eller salicylater) var forbundet med en signifikant forværring af depression blandt en undergruppe af studiedeltagere [249,250].
I det første forsøg, der involverede 1,258 deprimerede patienter behandlet med citalopram i 12 uger, var frekvensen af remission signifikant lavere blandt dem, der havde taget NSAID'er mindst én gang i forhold til dem, der ikke havde taget (45 % versus 55 %, OR 0.64, P = 0.0002) [249]. Det andet forsøg, der involverede 1,545 MDD-personer, viste, at frekvensen af behandlingsresistent depression var signifikant højere blandt dem, der tog NSAID'er (ELLER 1.55, 95 % CI 1.21 til 2.00) [231]. Forværringen af depression i NSAID-grupperne er muligvis ikke mekanisk forbundet med NSAID-behandling, men i stedet relateret til samtidige kroniske medicinske tilstande [10,12-18], som nødvendiggør langvarige NSAID'er, og som vides at være uafhængigt forbundet med øget risiko for behandlingsresistent depression [249,251]. Fremtidige undersøgelser, der undersøger virkningen af NSAID'er på depression og respons på antidepressiva hos mennesker, er nødvendige.
I andre eksperimentelle undersøgelser, der anvender akut-stress-paradigmer til at inducere en depressionslignende tilstand hos mus, øgede citalopram TNF-a, IFN-a og p11 (molekylær faktor forbundet med depressiv adfærd hos dyr) i frontal cortex, mens NSAID ibuprofen reducerede disse molekyler; NSAID'er svækkede også de antidepressive virkninger af SSRI'er, men ikke andre antidepressiva [249]. Disse resultater tyder på, at proinflammatoriske cytokiner paradoksalt nok kan udøve antidepressive virkninger på trods af overvældende beviser fra�humane undersøgelser om det modsatte (som gennemgået ovenfor), som kan svækkes af NSAID'er [249]. Mindst to overvejelser kan forklare dette tilsyneladende paradoks: 1) under nogle eksperimentelle forhold er proinflammatoriske cytokiner blevet forbundet med en neurobeskyttende rolle, [251; (for�for eksempel IFN-? i lave niveauer kan inducere neurobeskyttende mikroglia (figur 2) [163,166,251]); og 2) hvorvidt disse reaktioner observeret i forbindelse med et akut stressparadigme i en dyremodel er anvendelige på endogen MDD hos mennesker, forbliver uklart [251].
De terapeutiske virkninger af COX-2-hæmmere i psykiatriske lidelser kan involvere modulering af biosyntese af COX-2-afledte prostaglandiner, herunder proinflammatorisk PGE2 og antiinflammatorisk 15-deoxy-?12,14-PGJ2 (15d-PGJ2) [252,253]. COX-2-hæmmere kan reducere PGE2-medieret inflammation, hvilket kan bidrage til patofysiologien af psykiatriske lidelser [252,253]. De kan også ændre niveauerne af 15d-PGJ2 og aktiviteten af dets nuklear receptor peroxisom proliferator-aktiverede nuklear receptor gamma (PPAR-?) [252,253].
Adskillige undersøgelser tyder på, at 15d-PGJ2 og dets nukleare receptor PPAR-? kan tjene som biologiske markører for skizofreni [253]. Hos skizofrene patienter er serum-PGE2-niveauer øget, hvorimod serumniveauer af 15d-PGJ2 er faldet, ligesom ekspressionen af dets nukleare receptor PPAR-? i PBMC [252]. Mens COX-2-hæmmere kan begrænse de potentielt gavnlige antiinflammatoriske virkninger af den COX-2�afhængige �15d-PGJ2/PPAR-? kan de med fordel reducere dens skadelige virkninger, herunder 1) den øgede risiko for myokardieinfarkt og visse infektioner (f.eks. cytomegalovirus og Toxoplasma gondii) hos skizofrene patienter [254] og 2) dets pro-apoptotiske virkninger observeret i humant og animalsk cancervæv [255]. Andre potentielle mekanismer af COX-2-hæmmeres terapeutiske virkninger kan involvere deres evne til at reducere proinflammatoriske cytokinniveauer [163], begrænse quinolinsyreexcitotoksicitet (som i MDD) og sænke KYNA-niveauer (som ved skizofreni) [128].
Minocyclin kan være effektivt ved psykiatriske lidelser (tabel 3) [248]. In vitro-data tyder på, at minocyclin hæmmer MAP, cytokinsekretion, COX-2/PGE-2-ekspression og inducerbar nitrogenoxidsyntase [256]. Minocyclin kan også modvirke dysreguleret glutamaterg og dopaminerg neurotransmission [256].
Omega-3-fedtsyreeffektivitet ved psykiatriske lidelser er uklar [248]. I en metaanalyse fra 2011 af 15 randomiserede kontrollerede undersøgelser (916 MDD) reducerede omega-3-tilskud indeholdende eicosapentaensyre ?60 % (dosisområde 200 til 2,200 mg/d ud over dosis af docosahexaensyre) signifikant reduceret terapien til en depressiv (S.0.001)-symptomer til <.246cRI (P). 3]. En efterfølgende metaanalyse konkluderede imidlertid, at der ikke er nogen signifikant fordel ved omega-247 fedtsyrer ved depression, og at den påståede effekt blot er et resultat af publikationsbias [2012]. En metaanalyse fra 5 af 291 randomiserede-kontrollerede forsøg inklusive 3 BPD-deltagere fandt, at depressive, men ikke maniske, symptomer var signifikant forbedret blandt dem, der var randomiseret til omega-0.34 fedtsyrer i forhold til dem, der fik placebo (Hedges g 0.025, P = 257) [12]. I et randomiseret kontrolleret forsøg med skizofrene forsøgspersoner fulgt op til 66 måneder, var både positive og negative symptomscores signifikant faldet blandt de 3 deltagere randomiseret til langkædet omega-1.2 (12 g/dag i 0.02 uger; henholdsvis P = 0.01 og 258) [XNUMX]; den�Forfattere konkluderede, at omega-3-forøgelse under det tidlige forløb af skizofreni også kan forhindre tilbagefald og sygdomsprogression [258].
En metaanalyse fra 2012 af syv randomiserede kontrollerede forsøg, der vurderede omega-3-forøgelse hos 168 skizofrene patienter, fandt ingen fordel ved behandling [259]. Forfatterne af denne meta-analyse erklærede specifikt, at der ikke kunne drages nogen konklusion vedrørende endepunkterne for tilbagefaldsforebyggelse eller sygdomsprogression [259]. Eksperimentelle data tyder på, at eicosapentaensyre og docosahexaensyre medierer deres antiinflammatoriske virkninger ved at fremme syntesen af resolviner og protectiner, som kan hæmme leukocytinfiltration og reducere cytokinproduktion [248].
Neurosteroider, herunder pregnenolon og dets nedstrømsmetabolit allopregnanolon, kan have en gavnlig rolle i nogle psykiatriske lidelser [248,260]. I MDD fandt adskillige undersøgelser nedsatte plasma/CSF-allopregnanolon-niveauer korreleret med symptomsværhedsgrad, som normaliserede sig efter vellykket behandling med visse antidepressiva (f.eks. SSRI'er) og elektrokonvulsiv terapi [261]. Ved skizofreni kan prægnenolonniveauer i hjernen ændres [248], og serumallopregnanolonniveauer kan stige efter nogle antipsykotiske lægemidler (f.eks. clozapin og olanzapin) [260]. I tre randomiserede-kontrollerede forsøg (100 skizofreni (samlet); behandlingsvarighed, ca. ni uger) blev positive, negative og kognitive symptomer, såvel som ekstrapyramidale bivirkninger af antipsykotika signifikant forbedret i et eller flere forsøg blandt dem, der var randomiseret til pregnenolon i forhold til dem, der fik placebo [248]. I et forsøg blev forbedringen opretholdt med langvarig behandling med pregnenolon [248]. Pregnenolon kan regulere kognition og adfærd ved at forstærke funktionen af NMDA- og GABAA-receptorer [248]. Desuden kan allopregnanolon udøve neurobeskyttende og antiinflammatoriske virkninger [248]. Flere RCT-undersøgelser er nødvendige for at bekræfte den gavnlige rolle, neuroaktive steroider spiller i tidligt opståede psykiatriske lidelser hos mennesker.
Vi afventer resultaterne af flere igangværende kliniske forsøg, der undersøger de terapeutiske virkninger af andre antiinflammatoriske midler, herunder salicylat, en hæmmer af NF-?B (NCT01182727); acetylsalicylsyre (NCT01320982); pravastatin (NCT1082588); og dextromethorphan, en ikke-kompetitiv NMDAR-antagonist, der kan begrænse inflammationsinduceret dopaminerg neuronal skade (NCT01189006).
Fremtidige behandlingsstrategier
Selvom nuværende immunterapier (f.eks. IVIG, plasmaferese, kortikosteroider og immunsuppressive midler) ofte er effektive til behandling af autoimmune encephalitider, hvor inflammation er akut, intens og overvejende af adaptiv oprindelse, er deres effektivitet ved klassiske psykiatriske lidelser, hvor inflammation er kronisk,�meget mildere, og overvejende af medfødt oprindelse, er begrænset [2]. Udvikling af nye terapeutiske midler bør sigte mod at vende gliatab [46,138], nedregulere skadelig MAP, samtidig med at endogene neurobeskyttende T-regs og gavnligt MAP optimeres, snarere end vilkårligt at undertrykke inflammation, som det sker med nuværende immunsuppressive midler. Derudover er der behov for udvikling af potente co-adjuvans antioxidanter, der ville vende oxidativ skade ved psykiatriske lidelser.
konklusioner
Autoimmunitet kan forårsage et væld af neuropsykiatriske lidelser, der i begyndelsen kan vise sig med isolerede psykiatriske symptomer. Medfødt inflammation/autoimmunitet kan være relevant for patogenesen af psykiatriske symptomer hos en undergruppe af patienter med klassiske psykiatriske lidelser. Medfødt inflammation kan være mekanisk forbundet med de traditionelle monoaminerge og glutamaterge abnormiteter og øget oxidativ skade rapporteret i psykiatriske sygdomme.
Souhel Najjar1,5*, Daniel M Pearlman2,5, Kenneth Alper4, Amanda Najjar3 og Orrin Devinsky1,4,5
Forkortelser
3-OH-KYN: 3-hydroxy-kynurenin; a7nAchR: Alpha 7 nikotiniske acetylcholinreceptorer; AMPAR: Amino-3-hydroxy-5-methyl-l-4-isoxazolpropionsyrereceptorer; APC: Antigenpræsenterende celle; BBB: Blod-hjerne-barriere;
BH4: Tetrahydrobiopterin; BPD: Bipolar lidelse; CI: Konfidensinterval;
CNS: Centralnervesystemet; COX-2: Cyclooxegenase-2; CSF: Cerebrospinalvæske; DSM-IV: Diagnostic and Statistical Manual of Mental Disorders 4. udgave; EAAT'er: Excitatoriske aminosyretransportører; eNOS: Endothelial nitrogenoxidsyntase; GABAB: Gamma-aminosmørsyre-beta; GAD: glutaminsyredecarboxylase; GFAP: Glialt fibrillært surt protein; GLX: 1H MRS påviselig glutamat, glutamin, gamma-aminosmørsyre-komposit;
IDO: Indolamin 2,3-dioxygenase; Ig: Immunoglobulin; IL: Interleukin; IL-1RA: Interleukin 1-receptorantagonist; IFN-a: Interferon gamma;
KAT: Kynureninaminotransferase; KMO: Kynurenin 3-monooxygenase; KYN: Kynurenin; KYNA: Kynurensyre; LE: Limbisk encephalitis;
LPS: Lipopolysaccharid; MAP: Mikroglial aktivering og spredning;
MDD: Major depressiv lidelse; mGluR: Metabotropisk glutamatreceptor; MHC: II Major histokompatibilitetskompleks klasse to; MRI: Magnetisk resonansbilleddannelse; MRS: Magnetisk resonansspektroskopi; NF-AB: Nuklear faktor kappa B; NMDAR: N-methyl-D-aspartatreceptor; NR1: Glycinsted;
OCD: Obsessiv-kompulsiv lidelse; ELLER: Oddsforhold; PANDAS: Pædiatriske neuropsykiatriske autoimmune lidelser forbundet med streptokokinfektioner; PBMC: Mononukleære celler fra perifert blod; PET: Positron-emissionstomografi; PFC: Præfrontal cortex; PGE-2: Prostaglandin E2; PPAR-
?: Peroxisomproliferator-aktiveret nuklear receptor gamma; QA: Quinolinsyre; RNS: Reaktive nitrogenarter; ROS: Reaktive oxygenarter;
sIL: Opløseligt interleukin; SLE: Systemisk lupus erythematosus; SRI: Serotonin-genoptagelseshæmmer; TNF-a: Tumornekrosefaktor alfa; T-regs: CD4+CD25+FOXP3+ T-regulatoriske celler; TDO: Tryptophan-2,3-dioxygenase; Th: T-hjælper; VGKC: Spændingsstyret kaliumkanal; XAG-: Glutamataspartattransportør; Xc-: Natrium-uafhængig astroglial glutamat/cystin
antiporter system
Konkurrerende interesser
Forfatterne erklærer, at de ikke har nogen konkurrerende interesser.
Forfattere��Bidrag
SN og DMP udførte en omfattende litteraturgennemgang, fortolkede data, udarbejdede manuskript, figurer og tabeller. KA udarbejdede afsnittet om oxidative mekanismer og bidrog til manuskriptrevisionerne. AN og OD har kritisk revideret og forbedret manuskriptets design og kvalitet. Alle forfattere læste og godkendte det endelige manuskript.
Tak
Vi takker taknemmeligt for Dr. Josep Dalmau, MD, PhD, Tracy Butler, MD, og David Zazag, MD, PhD, for at levere deres ekspertise inden for henholdsvis autoimmune encephalitider, neuroinflammationsbilleddannelse og neuropatologi.
Forfatter�Detaljer
1Department of Neurology, New York University School of Medicine, 550 First Avenue, New York, NY 10016, USA. 2Geisel School of Medicine i Dartmouth, The Dartmouth Institute for Health Policy and Clinical Practice, 30 Lafayette Street, HB 7252, Lebanon, NH 03766, USA. 3Department of Pathology, Division of Neuropathology, New York University School of Medicine, 550 First Avenue, New York, NY 10016, USA. 4 Institut for Psykiatri, New York University School of Medicine, New York, NY, USA. 5New York University Comprehensive Epilepsy Center, 550 First Avenue, New York, NY 10016, USA.
Blank
Referencer:
1. Kayser MS, Dalmau J: Den nye forbindelse mellem autoimmune lidelser
og neuropsykiatrisk sygdom. J Neuropsychiatry Clin Neurosci 2011, 23:90-97.
2. Najjar S, Pearlman D, Zagzag D, Golfinos J, Devinsky O: Glutaminsyre
decarboxylase autoantistof syndrom, der viser sig som skizofreni.
Neurolog 2012, 18:88�91.
3. Graus F, Saiz A, Dalmau J: Antistoffer og neuronal autoimmun
lidelser i CNS. J Neurol 2010, 257:509�517.
4. Lennox BR, Coles AJ, Vincent A: Antistof-medieret encephalitis: a
behandlingsbar årsag til skizofreni. Br J Psychiatry 2012, 200:92�94.
5. Zandi MS, Irani SR, Lang B, Waters P, Jones PB, McKenna P, Coles AJ, Vincent
A, Lennox BR: Sygdomsrelevante autoantistoffer i første episode
skizofreni. J Neurol 2011, 258:686-688.
6. Bataller L, Kleopa KA, Wu GF, Rossi JE, Rosenfeld MR, Dalmau J:
Autoimmun limbisk encephalitis hos 39 patienter: immunfænotyper og
resultater. J Neurol Neurosurg Psychiatry 2007, 78:381-385.
7. Dale RC, Heyman I, Giovannoni G, Church AW: Forekomst af anti-hjerne
antistoffer hos børn med obsessiv-kompulsiv lidelse. Br J Psykiatri
2005, 187:314�319.
8. Kendler KS: Den plettede natur af årsager til psykiatrisk sygdom: erstatning
den organisk-funktionelle/hardware-software dikotomi med empirisk
baseret pluralisme. Mol Psykiatri 2012, 17:377�388.
9. Keskin G, Sunter G, Midi I, Tuncer N: Neurosyfilis som årsag til kognitiv
tilbagegang og psykiatriske symptomer i yngre alder. J Neuropsykiatri Clin
Neurosci 2011, 23:E41�E42.
10. Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R, Kupfer DJ: Can
bipolar lidelse ses som en multi-system inflammatorisk sygdom?
J Affect Disord 2012, 141:1�10.
11. Hackett ML, Yapa C, Parag V, Anderson CS: Hyppighed af depression efter
slagtilfælde: en systematisk gennemgang af observationsstudier. Slagtilfælde 2005, 36:1330�1340.
12. Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW: Fra
betændelse til sygdom og depression: når immunsystemet
undertvinger hjernen. Nat Rev Neurosci 2008, 9:46-56.
13. Laske C, Zank M, Klein R, Stransky E, Batra A, Buchkremer G, Schott K:
Autoantistofreaktivitet i serum fra patienter med svær depression,
skizofreni og sunde kontroller. Psychiatry Res 2008, 158:83-86.
14. Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR:
Inflammationsinduceret anhedoni: endotoksin reducerer ventral striatum
svar på belønning. Biol Psychiatry 2010, 68:748-754.
15. Haroon E, Raison CL, Miller AH: Psykoneuroimmunologi møder
neuropsykofarmakologi: translationelle implikationer af virkningen af
betændelse i adfærd. Neuropsychopharmacology 2012, 37:137-162.
16. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB:
Autoimmune sygdomme og alvorlige infektioner som risikofaktorer for
skizofreni: en 30-årig befolkningsbaseret registerundersøgelse. Am J Psykiatri
2011, 168:1303�1310.
17. McNally L, Bhagwagar Z, Hannestad J: Inflammation, glutamat og glia
i depression: en litteraturgennemgang. CNS Spectr 2008, 13:501�510.
18. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD:
Inflammation forårsager humørsvingninger gennem ændringer i subgenual
cingulat aktivitet og mesolimbisk forbindelse. Biol Psykiatri 2009,
66:407�414.19. Raison CL, Miller AH: Er depression en inflammatorisk lidelse?
Curr Psychiatry Rep 2011, 13:467�475.
20. Raison CL, Miller AH: Den evolutionære betydning af depression i
Patogen Host Defense (PATHOS-D). Mol Psykiatri 2013, 18:15�37.
21. Steiner J, Bogerts B, Sarnyai Z, Walter M, Gos T, Bernstein HG, Myint AM:
At bygge bro mellem immun- og glutamathypoteserne
skizofreni og svær depression: Glial NMDAs potentielle rolle
receptormodulatorer og svækket blodhjernebarriereintegritet. Verden J
Biol Psychiatry 2012, 13:482�492.
22. Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, Bogerts B:
Fordeling af HLA-DR-positive mikroglia i skizofreni afspejler
nedsat cerebral lateralisering. Acta Neuropathol 2006, 112:305-316.
23. Papakostas GI, Shelton RC, Kinrys G, Henry ME, Bakow BR, Lipkin SH, Pi B,
Thurmond L, Bilello JA: Vurdering af en multi-assay, serum-baseret
biologisk diagnostisk test for svær depressiv lidelse: en pilot og
replikationsundersøgelse. Mol Psykiatri 2013, 18:332�339.
24. Krishnan R: Unipolar depression hos voksne: epidemiologi, patogenese og
neurobiologi. I UpToDate. Redigeret af Basow DS. Waltham, MA: UpToDate; 2013.
25. Stovall J: Bipolar lidelse hos voksne: epidemiologi og diagnose. I
Opdateret. Redigeret af Basow DS. UpToDate: Waltham; 2013.
26. Fischer BA, Buchanan RW: Skizofreni: epidemiologi og patogenese.
I UpToDate. Redigeret af Basow DS. Waltham, MA: UpToDate; 2013.
27. Nestadt G, Samuels J, Riddle M, Bienvenu OJ 3rd, Liang KY, LaBuda M,
Walkup J, Grados M, Hoehn-Saric R: En familieundersøgelse af obsessiv-kompulsiv
sygdom. Arch Gen Psychiatry 2000, 57:358-363.
28. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D,
Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O,
Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J,
Lonnqvist J, Paunio T, Bürglum AD, Hartmann A, Fink-Jensen A, Nordentoft
M, Hougaard D, Norgaard-Pedersen B, B�ttcher Y, Olesen J, Breuer R, M�ller
HJ, Giegling I, et al: Almindelige varianter, der giver risiko for skizofreni.
Nature 2009, 460:744�747.
29. Müller N, Schwarz MJ: Den immunmedierede ændring af serotonin og
glutamat: mod et integreret syn på depression. Mol Psykiatri
2007, 12:988�1000.
30. Galecki P, Florkowski A, Bienkiewics M, Szemraj J: Funktionel polymorfi
af cyclooxygenase-2-genet (G-765C) hos depressive patienter.
Neuropsychobiology 2010, 62:116-120.
31. Levinson DF: Depressionens genetik: en gennemgang. Biol Psychiatry 2006,
60: 84 92.
32. Zhai J, Cheng L, Dong J, Shen Q, Zhang Q, Chen M, Gao L, Chen X, Wang K,
Deng X, Xu Z, Ji F, Liu C, Li J, Dong Q, Chen C: S100B-gen
polymorfismer forudsiger præfrontal rumlig funktion i både skizofreni
patienter og raske personer. Schizophr Res 2012, 134:89�94.
33. Zhai J, Zhang Q, Cheng L, Chen M, Wang K, Liu Y, Deng X, Chen X, Shen Q,
Xu Z, Ji F, Liu C, Dong Q, Chen C, Li J: Risikovarianter i S100B-genet,
forbundet med forhøjede S100B-niveauer, er også forbundet med
visuospatial handicap af skizofreni. Behav Brain Res 2011, 217:363�368.
34. Cappi C, Muniz RK, Sampaio AS, Cordeiro Q, Brentani H, Palacios SA,
Marques AH, Vallada H, Miguel EC, Guilherme L, Hounie AG: Association
undersøgelse mellem funktionelle polymorfier i TNF-alfa-genet og
tvangslidelse. Arq Neuropsiquiatr 2012, 70:87�90.
35. Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY,
Stockmeier CA, Rajkowska G: Glialt fibrillært surt protein
immunreaktivitet i den præfrontale cortex adskiller yngre fra
ældre voksne med svær depressiv lidelse. Biol Psychiatry 2000, 48:861-873.
36. Altshuler LL, Abulseoud OA, Foland Ross L, Bartzokis G, Chang S, Mintz J,
Hellemann G, Vinters HV: Amygdala astrocytreduktion hos forsøgspersoner med
svær depressiv lidelse, men ikke bipolar lidelse. Bipolar lidelse 2010,
12: 541 549.
37. Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH:
Immunhistokemisk lokalisering af phosphoryleret glialfibrillær sur
protein i den præfrontale cortex og hippocampus fra patienter med
skizofreni, bipolar lidelse og depression. Brain Behav Immun 2001,
15: 388 400.
38. Doyle C, Deakin JFW: Færre astrocytter i frontal cortex ved skizofreni,
depression og bipolar lidelse. Schizophrenia Res 2002, 53:106.
39. Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey
EF, Yolken RH: Sygdomsspecifikke ændringer i frontale cortex hjerneproteiner
ved skizofreni, bipolar lidelse og svær depressiv lidelse, The
Stanley Neuropathology Consortium. Mol Psykiatri 2000, 5:142-149.
40. Gosselin RD, Gibney S, O'Malley D, Dinan TG, Cryan JF: Regionsspecifik
fald i glial fibrillært surt protein immunreaktivitet i hjernen af
en rottemodel af depression. Neuroscience 2009, 159:915-925.
41. Banasr M, Duman RS: Glialtab i den præfrontale cortex er tilstrækkelig til at
fremkalde depressiv-lignende adfærd. Biol Psychiatry 2008, 64:863-870.
42. Cotter D, Hudson L, Landau S: Beviser for orbitofrontal patologi i
bipolar lidelse og svær depression, men ikke ved skizofreni.
Bipolar lidelse 2005, 7:358-369.
43. Brauch RA, Adnan El-Masri M, Parker Jr., El-Mallakh RS: Glialcellenummer
og neuron/gliacelleforhold i postmortem hjerner hos bipolære individer.
J Affect Disord 2006, 91:87�90.
44. Cotter DR, Pariante CM, Everall IP: Glialcelleabnormiteter i større
Psykiatriske lidelser: beviser og implikationer. Brain Res Bull 2001,
55: 585 595.
45. Cotter D, Mackay D, Landau S, Kerwin R, Everall I: Reduceret gliacelletæthed
og neuronal størrelse i den forreste cingulate cortex i svær depressiv
sygdom. Arch Gen Psychiatry 2001, 58:545-553.
46. Bowley MP, Drevets WC, Ong�r D, Pris JL: Lave glialtal i
amygdala ved svær depressiv lidelse. Biol Psychiatry 2002, 52:404�412.
47. Toro CT, Hallak JE, Dunham JS, Deakin JF: Glialt fibrillært surt protein og
glutaminsyntetase i subregioner af præfrontal cortex ved skizofreni
og humørforstyrrelser. Neurosci Lett 2006, 404:276-281.
48. Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J,
Stockmeier C: Lagspecifikke reduktioner i GFAP-reaktive astroglia i
dorsolateral præfrontal cortex ved skizofreni. Schizophr Res 2002, 57:127-138.
49. Steffek AE, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH: Cortical
ekspression af glial fibrillært surt protein og glutaminsyntetase er
nedsat i skizofreni. Schizophr Res 2008, 103:71-82.
50. Damadzic R, Bigelow LB, Krimer LS, Goldenson DA, Saunders RC, Kleinman
JE, Herman MM: En kvantitativ immunhistokemisk undersøgelse af astrocytter i
den entorhinale cortex ved skizofreni, bipolar lidelse og major
depression: fravær af signifikant astrocytose. Brain Res Bull 2001, 55:611�618.
51. Benes FM, McSparren J, Bird ED, SanGiovanni JP, Vincent SL: Underskud i
små interneuroner i præfrontale og cingulate cortex hos skizofrene
og skizoaffektive patienter. Arch Gen Psychiatry 1991, 48:996-1001.
52. Müller N, Schwarz MJ: Immunsystem og skizofreni. Curr Immunol
Rev 2010, 6:213–220.
53. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, Mawrin C,
Brisch R, Bielau H, Meyer Zu Schwabedissen L, Bogerts B, Myint AM: Alvorlig
depression er forbundet med øget mikroglial quinolinsyre i
subregioner af den anterior cingulate gyrus: bevis for en immunmoduleret
glutamaterg neurotransmission? J Neuroinflammation 2011, 8:94.
54. Vostrikov VM, Uranova NA, Orlovskaya DD: Underskud af perineuronal
oligodendrocytter i den præfrontale cortex ved skizofreni og humør
lidelser. Schizophr Res 2007, 94:273-280.
55. Rajkowska G, Miguel-Hidalgo JJ: Gliogenese og glialpatologi i
depression. CNS Neurol Disord Drug Targets 2007, 6:219�233.
56. Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI:
Oligodendroglial tæthed i den præfrontale cortex ved skizofreni og
humørsygdomme: en undersøgelse fra Stanley Neuropathology Consortium.
Schizophr Res 2004, 67:269-275.
57. Uranova N: Beskadigelse og tab af oligodendrocytter er afgørende i
patogenese af skizofreni og humørsygdomme (fundsform
postmortem undersøgelser). Neuropsychopharmacology 2004, 29:S33.
58. Uranova NA, Orlovskaya DD, Vostrikov VM, Rachmanova VI: Faldet
tæthed af oligodendrogliale satellitter af pyramidale neuroner i lag III in
den præfrontale cortex ved skizofreni og humørsygdomme. Schizophr Res
2002, 53:107.
59. Vostrikov VM, Uranova NA, Rakhmanova VI, Orlovskaia DD: Sænket
oligodendroglial celletæthed i den præfrontale cortex ved skizofreni.
Zh Nevrol Psikhiatr Im SS Korsakova 2004, 104:47�51.
60. Uranova NA, Zimina IS, Vikhreva OV, Krukov NO, Rachmanova VI, Orlovskaya
DD: Ultrastrukturel beskadigelse af kapillærer i neocortex i
skizofreni. World J Biol Psychiatry 2010, 11:567-578.
61. Hof PR, Haroutunian V, Friedrich VL Jr, Byne W, Buitron C, Perl DP, Davis KL:
Tab og ændret rumlig fordeling af oligodendrocytter i superior
frontal gyrus ved skizofreni. Biol Psychiatry 2003, 53:1075-1085.
62. Davis KL, Stewart DG, Friedman JI, Buchsbaum M, Harvey PD, Hof PR,
Buxbaum J, Haroutunian V: Hvidstofændringer i skizofreni:
bevis for myelin-relateret dysfunktion. Arch Gen Psychiatry 2003,
60:443�456.63. Flynn SW, Lang DJ, Mackay AL, Goghari V, Vavasour IM, Whittall KP, Smith
GN, Arango V, Mann JJ, Dwork AJ, Falkai P, Honer WG: Abnormiteter ved
myelinisering ved skizofreni påvist in vivo med MR og postmortem
med analyse af oligodendrocytproteiner. Mol Psykiatri 2003,
8: 811 820.
64. Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya
DD: Oligodendrocytpatologiens rolle i skizofreni. Int J
Neuropsychopharmacol 2007, 10:537-545.
65. Byne W, Kidkardnee S, Tatusov A, Yiannoulos G, Buchsbaum MS,
Haroutunian V: Skizofreni-associeret reduktion af neuronal og
oligodendrocyttal i den forreste primære thalamuskerne.
Schizophr Res 2006, 85:245-253.
66. Hamidi M, Drevets WC, Pris JL: Glialreduktion i amygdala i dur
depressiv lidelse skyldes oligodendrocytter. Biol Psychiatry 2004,
55: 563 569.
67. Bayer TA, Buslei R, Havas L, Falkai P: Beviser for aktivering af mikroglia i
patienter med psykiatriske sygdomme. Neurosci Lett 1999, 271:126-128.
68. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bernstein HG,
Bogerts B: Immunologiske aspekter i selvmordets neurobiologi:
forhøjet mikroglial tæthed ved skizofreni og depression er
forbundet med selvmord. J Psychiatr Res 2008, 42:151-157.
69. Rao JS, Harry GJ, Rapoport SI, Kim HW: Øget excitotoksicitet og
neuroinflammatoriske markører i postmortem frontal cortex fra bipolar
lidelsespatienter. Mol Psykiatri 2010, 15:384�392.
70. Bernstein HG, Steiner J, Bogerts B: Gliaceller i skizofreni:
patofysiologisk betydning og mulige konsekvenser for terapien.
Expert Rev Neurother 2009, 9:1059�1071.
71. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR:
Hæmatopoietisk oprindelse af patologisk pleje i Hoxb8 mutante mus.
Cell 2010, 141:775�785.
72. Antony JM: Grooming og dyrkning med microglia. Sci Signal 2010, 3:jc8.
73. Wonodi I, Stine OC, Sathyasaikumar KV, Roberts RC, Mitchell BD, Hong LE,
Kajii Y, Thaker GK, Schwarcz R: Nedreguleret kynurenin 3-
monooxygenase-genekspression og enzymaktivitet ved skizofreni
og genetisk association med skizofreni-endofenotyper. Arch Gen
Psychiatry 2011, 68:665�674.
74. Raison CL, Lowry CA, Rook GA: Inflammation, sanitet og
bestyrtelse: tab af kontakt med co-evolved, tolerogen
mikroorganismer og patofysiologi og behandling af major
depression. Arch Gen Psychiatry 2010, 67:1211�1224.
75. Drexhage RC, Hoogenboezem TH, Versnel MA, Berghout A, Nolen WA,
Drexhage HA: Aktivering af monocyt- og T-celle-netværk hos patienter
med bipolar lidelse. Brain Behav Immun 2011, 25:1206-1213.
76. Steiner J, Jacobs R, Panteli B, Brauner M, Schiltz K, Bahn S, Herberth M,
Westphal S, Gos T, Walter M, Bernstein HG, Myint AM, Bogerts B: Akut
skizofreni er ledsaget af reduceret T-celle og øget B-celle
immunitet. Eur Arch Psychiatry Clin Neurosci 2010, 260:509�518.
77. Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D: The
glutamat-baseret genetisk immunhypotese i obsessiv-kompulsiv
lidelse, En integrerende tilgang fra gener til symptomer.
Neurovidenskab 2010, 165:408�417.
78. Y�ksel C, Ong�r D: Magnetisk resonansspektroskopi undersøgelser af
glutamat-relaterede abnormiteter i humørsygdomme. Biol Psykiatri 2010,
68: 785 794.
79. Rao JS, Kellom M, Reese EA, Rapoport SI, Kim HW: Dysreguleret glutamat
og dopamintransportører i postmortem frontal cortex fra bipolar
og skizofrene patienter. J Affect Disord 2012, 136:63�71.
80. Bauer D, Gupta D, Harotunian V, Meador-Woodruff JH, McCullumsmith RE:
Unormalt udtryk af glutamattransporter og transporter
interagerende molekyler i præfrontal cortex hos ældre patienter med
skizofreni. Schizophr Res 2008, 104:108-120.
81. Matute C, Melone M, Vallejo-Illarramendi A, Conti F: Øget udtryk
af den astrocytiske glutamattransportør GLT-1 i den præfrontale cortex af
skizofrene. 2005, 49:451�455.
82. Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH: Udtryk af
excitatoriske aminosyretransporter transkripter i thalamus hos forsøgspersoner
med skizofreni. Am J Psychiatry 2001, 158:1393-1399.
83. McCullumsmith RE, Meador-Woodruff JH: Striatal excitatorisk aminosyre
transporter transkript ekspression ved skizofreni, bipolar lidelse,
og svær depressiv lidelse. Neuropsychopharmacology 2002,
26: 368 375.
84. Pittenger C, Bloch MH, Williams K: Glutamatabnormiteter i obsessiv
tvangslidelse: neurobiologi, patofysiologi og behandling.
Pharmacol Ther 2011, 132:314�332.
85. Hashimoto K: Glutamats nye rolle i patofysiologien af
svær depressiv lidelse. Brain Res Rev 2009, 61:105-123.
86. Hashimoto K, Sawa A, Iyo M: Forøgede niveauer af glutamat i hjerner fra
patienter med stemningslidelser. Biol Psychiatry 2007, 62:1310-1316.
87. Burbaeva G, Boksha IS, Turishcheva MS, Vorobyeva EA, Savushkina OK,
Tereshkina EB: Glutaminsyntetase og glutamatdehydrogenase i
den præfrontale cortex hos patienter med skizofreni. Prog
Neuropsychopharmacol Biol Psychiatry 2003, 27:675-680.
88. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R,
Shankar SK: Anti-hjerne autoantistoffer og ændret excitatorisk
neurotransmittere ved obsessiv-kompulsiv lidelse.
Neuropsychopharmacology 2009, 34:2489-2496.
89. Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL,
Krystal JH, Mason GF: Subtype-specifikke ændringer af gammaaminosmørsyre
syre og glutamat hos patienter med svær depression.
Arch Gen Psychiatry 2004, 61:705-713.
90. Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff
Pol HE: Glutamat i skizofreni: en fokuseret gennemgang og meta-analyse
af 1H-MRS undersøgelser. Schizophr Bull 2013, 39:120–129.
91. Liu Y, Ho RC, Mak A: Interleukin (IL)-6, tumornekrosefaktor alfa
(TNF-alfa) og opløselige interleukin-2-receptorer (sIL-2R) er forhøjede i
patienter med svær depressiv lidelse: en metaanalyse og metaregression.
J Affect Disord 2012, 139:230�239.
92. Brietzke E, Stabellini R, Grassis-Oliveira R, Lafer B: Cytokiner i bipolar
lidelse: nylige fund, skadelige virkninger, men løfter for fremtiden
terapeutiske midler. CNS Spectr 2011. www.cnsspectrums.com/aspx/
articledetail.aspx?articleid=3596.
93. Denys D, Fluitman S, Kavelaars A, Heijnen C, Westenberg H: Faldet
TNF-alfa- og NK-aktivitet ved obsessiv-kompulsiv lidelse.
Psychoneuroendocrinology 2004, 29:945-952.
94. Brambilla F, Perna G, Bellodi L, Arancio C, Bertani A, Perini G, Carraro C, Gava
F: Plasma interleukin-1 beta og tumor nekrose faktor koncentrationer i
obsessiv-kompulsive lidelser. Biol Psychiatry 1997, 42:976-981.
95. Fluitman S, Denys D, Vulink N, Schutters S, Heijnen C, Westenberg H:
Lipopolysaccharid-induceret cytokinproduktion i obsessiv-kompulsiv
lidelse og generaliseret social angst. Psykiatri
Res 2010, 178:313�316.
96. Janelidze S, Mattei D, Westrin A, Traskman-Bendz L, Brundin L: Cytokine
niveauer i blodet kan skelne selvmordsforsøgere fra deprimerede
patienter. Brain Behav Immun 2011, 25:335�339.
97. Postal M, Costallat LT, Appenzeller S: Neuropsykiatriske manifestationer i
systemisk lupus erythematosus: epidemiologi, patofysiologi og
ledelse. CNS Drugs 2011, 25:721�736.
98. Kozora E, Hanly JG, Lapteva L, Filley CM: Kognitiv dysfunktion i
systemisk lupus erythematosus: fortid, nutid og fremtid.
Arthritis Rheum 2008, 58:3286�3298.
99. Lancaster E, Martinez-Hernandez E, Dalmau J: Encephalitis og antistoffer mod
synaptiske og neuronale celleoverfladeproteiner. Neurology 2011, 77:179-189.
100. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon
R: Klinisk erfaring og laboratorieundersøgelser hos patienter med antiNMDAR
encephalitis. Lancet Neurol 2011, 10:63�74.
101. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell
JK, Dalmau J: Undersøgelse af LGI1 som antigen ved limbisk encephalitis
tidligere tilskrevet kaliumkanaler: en sagsserie. Lancet Neurol
2010, 9:776�785.
102. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E,
Wilson C, Jacobs D, Lai M, Walker RW, Graus F, Bataller L, Illa I, Markx S, Strauss
KA, Peles E, Scherer SS, Dalmau J: Undersøgelser af caspr2, et autoantigen af
encephalitis og neuromyotoni. Ann Neurol 2011, 69:303�311.
103. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, Friedman
D, Skeen MB, Grisold W, Kimura A, Ohta K, Iizuka T, Guzman M, Graus F,
Moss SJ, Balice-Gordon R, Dalmau J: Antistoffer mod GABA(B)-receptoren i
limbisk hjernebetændelse med anfald: tilfældeserier og karakterisering af
antigen. Lancet Neurol 2010, 9:67�76.
104. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine
JC, Liebers E, Kornblum C, Bien CG, Honnorat J, Wong S, Xu J, Contractor A,
Balice-Gordon R, Dalmau J: Antistoffer mod metabotropisk glutamat
receptor 5 i Ophelia-syndromet. Neurology 2011, 77:1698-1701.105. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ,
Galetta SL, Dichter M, Alavi A, Rosenfeld MR, Dalmau J: Behandlingsresponsiv
limbisk encephalitis identificeret ved neuropil antistoffer: MR og
PET korrelerer. Brain 2005, 128:1764-1777.
106. Tofaris GK, Irani SR, Cheeran BJ, Baker IW, Cader ZM, Vincent A:
Immunterapi-responsiv chorea som det præsenterende træk ved LGI1-
antistof encephalitis. Neurology 2012, 79:195-196.
107. Najjar S, Pearlman D, Najjar A, Ghiasian V, Zagzag D, Devinsky O:
Ekstralimbisk autoimmun encephalitis forbundet med glutaminsyre
decarboxylase-antistoffer: en underdiagnosticeret enhed? Epilepsi adfærd
2011, 21:306�313.
108. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig
LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N,
Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F,
Dalmau J: Behandling og prognostiske faktorer for langsigtet resultat i
patienter med anti-NMDA receptor encephalitis: en observationskohorte
undersøgelse. Lancet Neurol 2013, 12:157�165.
109. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK,
Rosenfeld MR, Balice-Gordon R, Lynch DR: Anti-NMDA-receptor
encephalitis: caseserier og analyse af virkningerne af antistoffer.
Lancet Neurol 2008, 7:1091�1098.
110. Graus F, Boronat A, Xifro X, Boix M, Svigelj V, Garcia A, Palomino A, Sabater
L, Alberch J, Saiz A: Den ekspanderende kliniske profil af anti-AMPA-receptor
encephalitis. Neurology 2010, 74:857�859.
111. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, Mata S, Kremens
D, Vitaliani R, Geschwind MD, Bataller L, Kalb RG, Davis R, Graus F, Lynch DR,
Balice-Gordon R, Dalmau J: AMPA-receptorantistoffer i limbisk
encephalitis ændre synaptisk receptor placering. Ann Neurol 2009, 65:424�434.
112. Najjar S, Pearlman D, Devinsky O, Najjar A, Nadkarni S, Butler T, Zagzag D:
Neuropsykiatrisk autoimmun encephalitis med negativt VGKC-kompleks,
NMDAR og GAD autoantistoffer: en caserapport og litteraturgennemgang,
kommende. Cogn Behav Neurol. I tryk.
113. Najjar S, Pearlman D, Zagzag D, Devinsky O: Spontant løsning
seronegativ autoimmun limbisk encephalitis. Cogn Behav Neurol 2011,
24: 99 105.
114. Gabilondo I, Saiz A, Galan L, Gonzalez V, Jadraque R, Sabater L, Sans A,
Sempere A, Vela A, Villalobos F, Vi�als M, Villoslada P, Graus F: Analyse af
tilbagefald i anti-NMDAR encephalitis. Neurology 2011, 77:996�999.
115. Barry H, Hardiman O, Healy DG, Keogan M, Moroney J, Molnar PP, Cotter
DR, Murphy KC: Anti-NMDA receptor encephalitis: en vigtig
differentialdiagnose ved psykose. Br J Psychiatry 2011, 199:508-509.
116. Dickerson F, Stallings C, Vaughan C, Origoni A, Khushalani S, Yolken R:
Antistoffer mod glutamatreceptoren ved mani. Bipolar lidelse 2012,
14: 547 553.
117. O'Loughlin K, Ruge P, McCauley M: Encephalitis og skizofreni: a
spørgsmål om ord. Br J Psychiatry 2012, 201:74.
118. Parratt KL, Allan M, Lewis SJ, Dalmau J, Halmagyi GM, Spies JM: Akut
Psykiatrisk sygdom hos en ung kvinde: en usædvanlig form for hjernebetændelse.
Med J Aust 2009, 191:284�286.
119. Suzuki Y, Kurita T, Sakurai K, Takeda Y, Koyama T: Sagsrapport om anti-NMDA
receptor encephalitis mistænkt for skizofreni. Seishin Shinkeigaku
Zasshi 2009, 111:1479-1484.
120. Tsutsui K, Kanbayashi T, Tanaka K, Boku S, Ito W, Tokunaga J, Mori A,
Hishikawa Y, Shimizu T, Nishino S: Anti-NMDA-receptor antistof påvist
ved encephalitis, skizofreni og narkolepsi med psykotiske træk.
BMC Psykiatri 2012, 12:37.
121. Van Putten WK, Hachimi-Idrissi S, Jansen A, Van Gorp V, Huyghens L:
Usædvanlig årsag til psykotisk adfærd hos en 9-årig pige: et tilfælde
rapport. Case Report Med 2012, 2012:358520.
122. Masdeu JC, Gonzalez-Pinto A, Matute C, Ruiz De Azua S, Palomino A, De
Leon J, Berman KF, Dalmau J: Serum IgG antistoffer mod NR1
underenhed af NMDA-receptoren ikke påvist ved skizofreni. Am J
Psychiatry 2012, 169:1120�1121.
123. Kirvan CA, Swedo SE, Kurahara D, Cunningham MW: Streptokokmimik
og antistof-medieret cellesignalering i patogenesen af
Sydenhams chorea. Autoimmunitet 2006, 39:21�29.
124. Swedo SE: Streptokokinfektion, Tourettes syndrom og OCD: er der
en forbindelse? Pandaer: Hest eller zebra? Neurology 2010, 74:1397-1398.
125. Morer A, Lazaro L, Sabater L, Massana J, Castro J, Graus F: Antineuronal
antistoffer i en gruppe børn med obsessiv-kompulsiv lidelse
og Tourettes syndrom. J Psychiatr Res 2008, 42:64-68.
126. Pavone P, Bianchini R, Parano E, Incorpora G, Rizzo R, Mazzone L, Trifiletti RR:
Anti-hjerne-antistoffer i PANDAS versus ukomplicerede streptokok
infektion. Pediatr Neurol 2004, 30:107�110.
127. Maina G, Albert U, Bogetto F, Borghese C, Berro AC, Mutani R, Rossi F,
Vigliani MC: Anti-hjerne-antistoffer hos voksne patienter med obsessiv-kompulsiv
sygdom. J Affect Disord 2009, 116:192�200.
128. Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, Klein J,
Moses AE, Somnier FE, Leckman JF, Swedo SE, Cunningham MW, Joel D:
Adfærdsmæssige, farmakologiske og immunologiske abnormiteter efter
streptokokeksponering: en ny rottemodel af Sydenham chorea og
relaterede neuropsykiatriske lidelser. Neuropsykofarmakologi 2012,
37: 2076 2087.
129. Dale RC, Candler PM, Church AJ, Wait R, Pocock JM, Giovannoni G:
Neuronale overfladeglykolytiske enzymer er autoantigen-mål i
post-streptokok autoimmun CNS-sygdom. J Neuroimmunol 2006,
172: 187 197.
130. Nicholson TR, Ferdinando S, Krishnaiah RB, Anhoury S, Lennox BR, MataixCols
D, Cleare A, Veale DM, Drummond LM, Fineberg NA, Church AJ,
Giovannoni G, Heyman I: Forekomst af anti-basale ganglia antistoffer i
obsessiv-kompulsiv lidelse for voksne: tværsnitsundersøgelse. Br J Psykiatri
2012, 200:381�386.
131. Wu K, Hanna GL, Rosenberg DR, Arnold PD: Glutamats rolle
signalering i patogenesen og behandlingen af obsessiv-kompulsiv
sygdom. Pharmacol Biochem Behav 2012, 100:726�735.
132. Perlmutter SJ, Leitman SF, Garvey MA, Hamburger S, Feldman E, Leonard
HL, Swedo SE: Terapeutisk plasmaudveksling og intravenøs
immunglobulin til obsessiv-kompulsiv lidelse og tic-lidelser i
barndom. Lancet 1999, 354:1153-1158.
133. Pereira A Jr, Furlan FA: Astrocytter og menneskelig kognition: modellering
informationsintegration og modulering af neuronal aktivitet.
Prog Neurobiol 2010, 92:405-420.
134. Barres BA: Glias mysterium og magi: et perspektiv på deres roller i
sundhed og sygdom. Neuron 2008, 60:430�440.
135. Verkhratsky A, Parpura V, Rodriguez JJ: Hvor tankerne bor:
fysiologi af neuronalt-glialt "diffust neuralt net". Brain Res Rev 2011,
66: 133 151.
136. Sofroniew MV: Molekylær dissektion af reaktiv astrogliose og glialar
dannelse. Trends Neurosci 2009, 32:638�647.
137. Hamilton NB, Attwell D: Exocyterer astrocytter virkelig neurotransmittere?
Nat Rev Neurosci 2010, 11:227-238.
138. Rajkowska G: Postmortem undersøgelser af humørsygdomme indikerer ændret
antallet af neuroner og gliaceller. Biol Psychiatry 2000, 48:766-777.
139. Coupland NJ, Ogilvie CJ, Hegadoren KM, Seres P, Hanstock CC, Allen PS:
Nedsat præfrontal Myo-inositol ved svær depressiv lidelse.
Biol Psychiatry 2005, 57:1526�1534.
140. Miguel-Hidalgo JJ, Overholser JC, Jurjus GJ, Meltzer HY, Dieter L, Konick L,
Stockmeier CA, Rajkowska G: Vaskulær og ekstravaskulær immunreaktivitet
for intercellulært adhæsionsmolekyle 1 i den orbitofrontale cortex af
personer med svær depression: aldersafhængige ændringer. J Affektuorden
2011, 132:422�431.
141. Miguel-Hidalgo JJ, Wei JR, Andrew M, Overholser JC, Jurjus G, Stockmeier
CA, Rajkowska G: Glia patologi i den præfrontale cortex i alkohol
afhængighed med og uden depressive symptomer. Biol Psychiatry 2002,
52: 1121 1133.
142. Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY,
Uylings HB, Friedman L, Rajkowska G: Cellulære ændringer i postmortem
hippocampus i svær depression. Biol Psychiatry 2004, 56:640-650.
143. Ong�r D, Drevets WC, Pris JL: Glial reduktion i den subgenuelle præfrontale
cortex ved humørsygdomme. Proc Natl Acad Sci USA 1998, 95:13290�13295.
144. Gittins RA, Harrison PJ: En morfometrisk undersøgelse af glia og neuroner i
anterior cingulate cortex i humørsygdom. J Affect Disord 2011,
133: 328 332.
145. Cotter D, Mackay D, Beasley C, Kerwin R, Everall I: Reduceret glialdensitet
og neuronalt volumen ved svær depressiv lidelse og skizofreni i
den forreste cingulate cortex [abstrakt]. Schizophrenia Res 2000, 41:106.
146. Si X, Miguel-Hidalgo JJ, Rajkowska G: GFAP-ekspression er reduceret i
dorsolateral præfrontal cortex ved depression. I Society for Neuroscience; 2003.
Neurovidenskab mødeplan: New Orleans; 2003.
147. Legutko B, Mahajan G, Stockmeier CA, Rajkowska G: Hvidstof-astrocytter
reduceres ved depression. I Society for Neuroscience. Neurovidenskabsmøde
Planlægger: Washington, DC; 2011.148. Edgar N, Sibille E: En formodet funktionel rolle for oligodendrocytter i
humørregulering. Transl Psychiatry 2012, 2:e109.
149. Rajkowska G, Halaris A, Selemon LD: Reduktioner i neuronale og gliale
tæthed karakteriserer den dorsolaterale præfrontale cortex i bipolar
sygdom. Biol Psychiatry 2001, 49:741-752.
150. Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP: Reduceret
neuronal størrelse og gliacelletæthed i område 9 af den dorsolaterale
præfrontal cortex hos personer med svær depressiv lidelse. Cereb Cortex
2002, 12:386�394.
151. Stark AK, Uylings HB, Sanz-Arigita E, Pakkenberg B: Glialcelletab i
anterior cingulate cortex, en underregion af den præfrontale cortex, i
personer med skizofreni. Am J Psychiatry 2004, 161:882-888.
152. Konopaske GT, Dorph-Petersen KA, Sweet RA, Pierri JN, Zhang W, Sampson
AR, Lewis DA: Effekt af kronisk antipsykotisk eksponering på astrocytter og
oligodendrocyttal i makakaber. Biol Psykiatri 2008,
63: 759 765.
153. Selemon LD, Lidow MS, Goldman-Rakic PS: Øget volumen og glial
tæthed i primat præfrontal cortex forbundet med kronisk
eksponering for antipsykotiske lægemidler. Biol Psychiatry 1999, 46:161-172.
154. Steiner J, Bernstein HG, Bielau H, Farkas N, Winter J, Dobrowolny H, Brisch R,
Gos T, Mawrin C, Myint AM, Bogerts B: S100B-immunopositiv glia er
forhøjet i paranoid sammenlignet med resterende skizofreni: a
morfometrisk undersøgelse. J Psychiatr Res 2008, 42:868-876.
155. Carter CJ: eIF2B og oligodendrocytoverlevelse: hvor natur og næring
mødes ved bipolar lidelse og skizofreni? Schizophr Bull 2007,
33: 1343 1353.
156. Hayashi Y, Nihonmatsu-Kikuchi N, Hisanaga S, Yu XJ, Tatebayashi Y:
Neuropatologiske ligheder og forskelle mellem skizofreni
og bipolar lidelse: en flowcytometrisk postmortem hjerneundersøgelse.
PLoS One 2012, 7:e33019.
157. Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD: Ultrastrukturel
ændringer af myeliniserede fibre og oligodendrocytter i det præfrontale
cortex i skizofreni: en postmortem morfometrisk undersøgelse.
Schizophr Res Treatment 2011, 2011:325789.
158. Torres-Platas SG, Hercher C, Davoli MA, Maussion G, Labonte B, Turecki
G, Mechawar N: Astrocytisk hypertrofi i anterior cingulate hvid
spørgsmål om deprimerede selvmord. Neuropsykofarmakologi 2011,
36: 2650 2658.
159. Pereira A Jr, Furlan FA: Om rollen som synkronisering for neuron-astrocyt
interaktioner og perceptuel bevidst bearbejdning. J Biol Phys 2009,
35: 465 480.
160. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A: Physiology of
mikroglia. Physiol Rev 2011, 91:461�553.
161. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A: Rollen
af mikroglia i den raske hjerne. J Neurosci 2011, 31:16064-16069.
162. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, Le
Charpentier T, Josserand J, Ali C, Vivien D, Collingridge GL, Lombet A, Issa L,
Rene F, Loeffler JP, Kavelaars A, Verney C, Mantz J, Gressens P: Aktivering af
mikrogliale N-methyl-D-aspartat-receptorer udløser betændelse og
neuronal celledød i den udviklende og modne hjerne. Ann Neurol
2012, 72:536�549.
163. Schwartz M, Shaked I, Fisher J, Mizrahi T, Schori H: Beskyttende
autoimmunitet mod fjenden indeni: bekæmpelse af glutamattoksicitet.
Trends Neurosci 2003, 26:297-302.
164. Pacheco R, Gallart T, Lluis C, Franco R: Glutamats rolle på T-celle
medieret immunitet. J Neuroimmunol 2007, 185:9-19.
165. Najjar S, Pearlman D, Miller DC, Devinsky O: Refraktær epilepsi forbundet
med mikroglial aktivering. Neurolog 2011, 17:249�254.
166. Schwartz M, Butovsky O, Bruck W, Hanisch UK: Mikroglial fænotype: er
forpligtelse reversibel? Trends Neurosci 2006, 29:68�74.
167. Wang F, Wu H, Xu S, Guo X, Yang J, Shen X: Makrofagmigrering
hæmmende faktor aktiverer cyclooxygenase 2-prostaglandin E2 i dyrkede
spinal mikroglia. Neurosci Res 2011, 71:210-218.
168. Zhang XY, Xiu MH, Song C, Chenda C, Wu GY, Haile CN, Kosten TA, Kosten
TR: Øget serum S100B i aldrig-medicineret og medicineret
skizofrene patienter. J Psychiatr Res 2010, 44:1236-1240.
169. Kawasaki Y, Zhang L, Cheng JK, Ji RR: Cytokinmekanismer af central
sensibilisering: distinkt og overlappende rolle af interleukin-1beta,
interleukin-6 og tumornekrosefaktor-alfa til regulering af synaptisk og
neuronal aktivitet i den overfladiske rygmarv. J Neurosci 2008,
28: 5189 5194.
170. Müller N, Schwarz MJ: Det immunologiske grundlag for glutamatergic
forstyrrelse i skizofreni: mod et integreret syn. J Neural
Transm Suppl 2007, 72:269�280.
171. Hestad KA, Tonseth S, Stoen CD, Ueland T, Aukrust P: Forhøjede plasmaniveauer
af tumornekrosefaktor alfa hos patienter med depression: normalisering
under elektrokonvulsiv behandling. J ECT 2003, 19:183-188.
172. Kubera M, Kenis G, Bosmans E, Zieba A, Dudek D, Nowak G, Maes M:
Plasmaniveauer af interleukin-6, interleukin-10 og interleukin-1 receptor
antagonist ved depression: sammenligning mellem den akutte tilstand og efter
eftergivelse. Pol J Pharmacol 2000, 52:237-241.
173. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B: Meta-analyse af
cytokinændringer i skizofreni: klinisk status og antipsykotisk
effekter. Biol Psychiatry 2011, 70:663�671.
174. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E: Inflammatorisk
cytokinændringer i skizofreni: en systematisk kvantitativ gennemgang.
Biol Psychiatry 2008, 63:801�808.
175. Reale M, Patruno A, De Lutiis MA, Pesce M, Felaco M, Di Giannantonio M, Di
Nicola M, Grilli A: Dysregulering af kemo-cytokinproduktion i
skizofrene patienter versus raske kontroller. BMC Neurosci 2011, 12:13.
176. Fluitman SB, Denys DA, Heijnen CJ, Westenberg HG: Afsky påvirker TNFalpha,
IL-6 og noradrenalin niveauer hos patienter med obsessiv-kompulsiv
sygdom. Psychoneuroendocrinology 2010, 35:906-911.
177. Konuk N, Tekin IO, Ozturk U, Atik L, Atasoy N, Bektas S, Erdogan A: Plasma
niveauer af tumornekrosefaktor-alfa og interleukin-6 i obsessiv
tvangslidelse. Mediators Inflamm 2007, 2007:65704.
178. Monteleone P, Catapano F, Fabrazzo M, Tortorella A, Maj M: Faldet
blodniveauer af tumornekrosefaktor-alfa hos patienter med obsessiv-kompulsiv
sygdom. Neuropsychobiology 1998, 37:182-185.
179. Marazziti D, Presta S, Pfanner C, Gemignani A, Rossi A, Sbrana S, Rocchi V,
Ambrogi F, Cassano GB: Immunologiske ændringer hos voksne obsessiv-kompulsiv
sygdom. Biol Psychiatry 1999, 46:810-814.
180. Zai G, Arnold PD, Burroughs E, Richter MA, Kennedy JL: Tumornekrose
faktor-alfa-genet er ikke forbundet med obsessiv-kompulsiv lidelse.
Psychiatr Genet 2006, 16:43.
181. Rodr�guez AD, Gonz�lez PA, Garc�a MJ, de la Rosa A, Vargas M, Marrero F:
Circadian variationer i proinflammatoriske cytokinkoncentrationer i akutte
myokardieinfarkt. Rev Esp Cardiol 2003, 56:555�560.
182. Oliver JC, Bland LA, Oettinger CW, Arduino MJ, McAllister SK, Aguero SM,
Favero MS: Cytokinkinetik i en in vitro fuldblodsmodel, der følger
en endotoksin udfordring. Lymphokine Cytokine Res 1993, 12:115-120.
183. Le T, Leung L, Carroll WL, Schibler KR: Regulering af interleukin-10-genet
udtryk: mulige mekanismer, der tegner sig for dens opregulering og for
modningsforskelle i dets ekspression af mononukleære blodceller.
Blood 1997, 89:4112�4119.
184. Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ:
Karakterisering af ekspressionen af NMDA-receptorer hos mennesker
astrocytter. PLoS One 2010, 5:e14123.
185. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B:
Kynurenin-vej ved svær depression: tegn på svækkelse
neurobeskyttelse. J Affect Disord 2007, 98:143�151.
186. Sanacora G, Treccani G, Popoli M: Mod en glutamathypotese om
depression: en spirende grænse for neuropsykofarmakologi til
humørforstyrrelser. Neuropharmacology 2012, 62:63�77.
187. Saleh A, Schroeter M, Jonkmanns C, Hartung HP, Modder U, Jander S: In
vivo MRI af hjernebetændelse i humant iskæmisk slagtilfælde. Brain 2004,
127: 1670 1677.
188. Tilleux S, Hermans E: Neuroinflammation og regulering af glial glutamat
optagelse ved neurologiske lidelser. J Neurosci Res 2007, 85:2059-2070.
189. Helms HC, Madelung R, Waagepetersen HS, Nielsen CU, Brodin B: In vitro
beviser for hypotesen om udstrømning af glutamat i hjernen: endotel i hjernen
celler co-dyrket med astrocytter viser en polariseret hjerne-til-blod
transport af glutamat. 2012, 60:882�893.
190. Leonard BE: Begrebet depression som en dysfunktion af immunforsvaret
system. Curr Immunol Rev 2010, 6:205-212.
191. Labrie V, Wong AH, Roder JC: Bidrag fra D-serin-vejen til
skizofreni. Neuropharmacology 2012, 62:1484-1503.
192. Gras G, Samah B, Hubert A, Leone C, Porcheray F, Rimaniol AC: EAAT
udtryk ved makrofager og mikroglia: stadig flere spørgsmål end
svar. Aminosyrer 2012, 42:221�229.
193. Livingstone PD, Dickinson JA, Srinivasan J, Kew JN, Wonnacott S:
Glutamat-dopamin krydstale i rottens præfrontale cortex moduleres af Alpha7 nikotinreceptorer og forstærkes af PNU-120596. J Mol
Neurosci 2010, 40:172–176.194. Kondziella D, Brenner E, Eyjolfsson EM, Sonnewald U: How do glialneuronal
interaktioner passer ind i aktuelle neurotransmitterhypoteser om
skizofreni? Neurochem Int 2007, 50:291�301.
195. Wu HQ, Pereira EF, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R: The
astrocyt-afledt alpha7 nikotinreceptorantagonist kynurensyre
kontrollerer ekstracellulære glutamatniveauer i den præfrontale cortex. J Mol
Neurosci 2010, 40:204-210.
196. Steiner J, Bogerts B, Schroeter ML, Bernstein HG: S100B protein i
neurodegenerative lidelser. Clin Chem Lab Med 2011, 49:409�424.
197. Steiner J, Marquardt N, Pauls I, Schiltz K, Rahmoune H, Bahn S, Bogerts B,
Schmidt RE, Jacobs R: Humane CD8(+) T-celler og NK-celler udtrykker og
udskiller S100B ved stimulering. Brain Behav Immun 2011, 25:1233-1241.
198. Shanmugam N, Kim YS, Lanting L, Natarajan R: Regulering af
cyclooxygenase-2 ekspression i monocytter ved ligering af receptoren
til avancerede glykeringsslutprodukter. J Biol Chem 2003, 278:34834-34844.
199. Rothermundt M, Ohrmann P, Abel S, Siegmund A, Pedersen A, Ponath G,
Suslow T, Peters M, Kaestner F, Heindel W, Arolt V, Pfleiderer B: Glialcelle
aktivering i en undergruppe af patienter med skizofreni angivet ved
øgede S100B serumkoncentrationer og forhøjet myo-inositol.
Prog Neuropsychopharmacol Biol Psychiatry 2007, 31:361-364.
200. Falcone T, Fazio V, Lee C, Simon B, Franco K, Marchi N, Janigro D: Serum
S100B: en potentiel biomarkør for suicidalitet hos unge? PLoS One
2010, 5:e11089.
201. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: Serum
markører understøtter sygdomsspecifik gliapatologi ved svær depression.
J Affect Disord 2008, 111:271�280.
202. Rothermundt M, Ahn JN, Jorgens S: S100B i skizofreni: en opdatering.
Gen Physiol Biophys 2009, 28 Spec No Focus:F76�F81.
203. Schroeter ML, Abdul-Khaliq H, Krebs M, Diefenbacher A, Blasig IE: Neuronspecifik
enolase er uændret, mens S100B er forhøjet i serum af
patienter med skizofreni original forskning og meta-analyse.
Psychiatry Res 2009, 167:66-72.
204. Rothermundt M, Missler U, Arolt V, Peters M, Leadbeater J, Wiesmann M,
Rudolf S, Wandinger KP, Kirchner H: Forøgede S100B-blodniveauer i
umedicinerede og behandlede skizofrene patienter er korreleret med
negativ symptomatologi. Mol Psychiatry 2001, 6:445-449.
205. Suchankova P, Klang J, Cavanna C, Holm G, Nilsson S, Jonsson EG, Ekman A:
Er Gly82Ser-polymorfien i RAGE-genet relevant for
skizofreni og personlighedstrækket psykoticisme? J Psykiatri Neurosci
2012, 37:122�128.
206. Scapagnini G, Davinelli S, Drago F, De Lorenzo A, Oriani G: Antioxidanter som
antidepressiva: fakta eller fiktion? CNS Drugs 2012, 26:477�490.
207. Ng F, Berk M, Dean O, Bush AI: Oxidativ stress i psykiatriske lidelser:
evidensgrundlag og terapeutiske implikationer. Int J Neuropsychopharmacol
2008, 11:851�876.
208. Salim S, Chugh G, Asghar M: Betændelse i angst. Adv Protein Chem
Struct Biol 2012, 88:1�25.
209. Anderson G, Berk M, Dodd S, Bechter K, Altamura AC, Dell'osso B, Kanba S,
Monji A, Fatemi SH, Buckley P, Debnath M, Das UN, Meyer U, M�ller N,
Kanchanatawan B, Maes M: Immuno-inflammatorisk, oxidativ og nitrosativ
stress og neuroprogressive veje i ætiologi, forløb og behandling
af skizofreni. Prog Neuropsychopharmacol Biol Psychiatry 2013, 42:1�42.
210. Coughlin JM, Ishizuka K, Kano SI, Edwards JA, Seifuddin FT, Shimano MA,
Daley EL, et al: Markant reduktion af opløselig superoxiddismutase-1
(SOD1) i cerebrospinalvæske hos patienter med nyligt debut
skizofreni. Mol Psykiatri 2012, 18:10�11.
211. Bombaci M, Grifantini R, Mora M, Reguzzi V, Petracca R, Meoni E, Balloni S,
Zingaretti C, Falugi F, Manetti AG, Margarit I, Musser JM, Cardona F, Orefici
G, Grandi G, Bensi G: Proteinarray-profilering af tic-patientsera afslører en
bred vifte og forbedret immunrespons mod gruppe A
Streptococcus antigener. PLoS One 2009, 4:e6332.
212. Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L,
Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E: TNF-alfa
nedregulerer eNOS-ekspression og mitokondriel biogenese i fedt
og muskler fra overvægtige gnavere. J Clin Invest 2006, 116:2791�2798.
213. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B: Mitokondrier, oxidativ
stress og celledød. Apoptosis 2007, 12:913-922.
214. Shalev H, Serlin Y, Friedman A: Breaching the blood brain barrier as a gate
til psykiatrisk lidelse. Kardiovaskulær psykiatri Neurol 2009, 2009:278531.
215. Abbott NJ, Ronnback L, Hansson E: Astrocyt-endotelinteraktioner ved
blod-hjerne-barriere. Nat Rev Neurosci 2006, 7:41�53.
216. Bechter K, Reiber H, Herzog S, Fuchs D, Tumani H, Maxeiner HG:
Cerebrospinalvæskeanalyse i affektive og skizofrene spektrum
lidelser: identifikation af undergrupper med immunresponser og
blod-CSF barriere dysfunktion. J Psychiatr Res 2010, 44:321-330.
217. Harris LW, Wayland M, Lan M, Ryan M, Giger T, Lockstone H, Wuethrich I,
Mimmack M, Wang L, Kotter M, Craddock R, Bahn S: Den cerebrale
mikrovaskulatur i skizofreni: en laserfangst mikrodissektionsundersøgelse.
PLoS One 2008, 3:e3964.
218. Lin JJ, Mula M, Hermann BP: Afdækning af neuroadfærd
komorbiditeter af epilepsi i løbet af levetiden. Lancet 2012, 380:1180�1192.
219. Isingrini E, Belzung C, Freslon JL, Machet MC, Camus V: Fluoxetin effekt på
aorta nitrogenoxid-afhængig vasorelaxation i det uforudsigelige
kronisk mild stressmodel af depression hos mus. Psychosom Med 2012,
74: 63 72.
220. Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY, Shen YC: Effekten af
risperidonbehandling på superoxiddismutase ved skizofreni. J Clin
Psychopharmacol 2003, 23:128-131.
221. Lavoie KL, Pelletier R, Arsenault A, Dupuis J, Bacon SL: Sammenslutning mellem
klinisk depression og endotelfunktion målt ved underarm
hyperæmisk reaktivitet. Psychosom Med 2010, 72:20�26.
222. Chrapko W, Jurasz P, Radomski MW, Archer SL, Newman SC, Baker G, Lara N,
Le Melledo JM: Ændring af nedsatte plasma NO-metabolitter og
blodplade NO-syntaseaktivitet af paroxetin hos deprimerede patienter.
Neuropsychopharmacology 2006, 31:1286-1293.
223. Chrapko WE, Jurasz P, Radomski MW, Lara N, Archer SL, Le Melledo JM:
Nedsat blodpladenitrogenoxidsyntaseaktivitet og plasmanitrogenoxid
metabolitter i svær depressiv lidelse. Biol Psychiatry 2004, 56:129-134.
224. Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S: Opdatering om mekanisme
og katalytisk regulering i NO-syntaserne. J Biol Chem 2004,
279: 36167 36170.
225. Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, Tsai AL, Zweier
JL: Peroxynitrit inducerer ødelæggelse af tetrahydrobiopterin og
hæm i endothelial nitrogenoxidsyntase: overgang fra reversibel til
irreversibel enzymhæmning. Biochemistry 2010, 49:3129�3137.
226. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen
YR, Druhan LJ, Zweier JL: S-glutathionylering kobler eNOS og
regulerer dets cellulære og vaskulære funktion. Nature 2010, 468:1115�1118.
227. Szabo C, Ischiropoulos H, Radi R: Peroxynitrit: biokemi,
patofysiologi og udvikling af terapeutika. Nat Rev Drug Discov
2007, 6:662�680.
228. Papakostas GI, Shelton RC, Zajecka JM, Etemad B, Rickels K, Clain A, Baer L,
Dalton ED, Sacco GR, Schoenfeld D, Pencina M, Meisner A, Bottiglieri T,
Nelson E, Mischoulon D, Alpert JE, Barbee JG, Zisook S, Fava M: Lmethylfolat
som supplerende behandling for SSRI-resistent svær depression:
resultater af to randomiserede, dobbeltblindede, parallel-sekventielle forsøg. Am J
Psychiatry 2012, 169:1267�1274.
229. Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P,
Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM: 5-
methyltetrahydrofolat forbedrer hurtigt endotelfunktionen og
reducerer superoxidproduktion i menneskelige kar: virkninger på vaskulær
tetrahydrobiopterin tilgængelighed og endothelial nitrogenoxidsyntase
kobling. Oplag 2006, 114:1193�1201.
230. Masano T, Kawashima S, Toh R, Satomi-Kobayashi S, Shinohara M, Takaya T,
Sasaki N, Takeda M, Tawa H, Yamashita T, Yokoyama M, Hirata K: Fordelagtigt
effekter af eksogent tetrahydrobiopterin på venstre ventrikulær ombygning
efter myokardieinfarkt hos rotter: den mulige rolle af oxidativ stress
forårsaget af ukoblet endotel nitrogenoxidsyntase. Circ J 2008,
72: 1512 1519.
231. Alp NJ, Channon KM: Regulering af endothelial nitrogenoxidsyntase ved
tetrahydrobiopterin ved vaskulær sygdom. Arterioscler Thromb Vasc Biol 2004,
24: 413 420.
232. Szymanski S, Ashtari M, Zito J, Degreef G, Bogerts B, Lieberman J:
Gadolinium-DTPA forbedret gradient ekko magnetisk resonans scanner ind
første episode af psykose og kroniske skizofrene patienter.
Psychiatry Res 1991, 40:203-207.
233. Butler T, Weisholtz D, Isenberg N, Harding E, Epstein J, Stern E, Silbersweig
D: Neuroimaging af frontal-limbisk dysfunktion ved skizofreni og
epilepsi-relateret psykose: mod en konvergent neurobiologi.
Epilepsi Behav 2012, 23:113�122.234. Butler T, Maoz A, Vallabhajosula S, Moeller J, Ichise M, Paresh K, Pervez F,
Friedman D, Goldsmith S, Najjar S, Osborne J, Solnes L, Wang X, French J,
Thesen T, Devinsky O, Kuzniecky R, Stern E, Silbersweig D: Billedbehandling
betændelse hos en patient med epilepsi forbundet med antistoffer mod
glutaminsyredecarboxylase [abstrakt]. I Am Epilepsy Society Abstracts,
Bind 2. Baltimore: American Epilepsy Society; 2011:191.
235. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers
E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS:
Mikroglia-aktivering i nyligt debuterende skizofreni: en kvantitativ (R)-
[11C]PK11195 positron emission tomografi undersøgelse. Biol Psykiatri 2008,
64: 820 822.
236. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC:
Neuroinflammation i skizofreni-relateret psykose: en PET-undersøgelse.
J Nucl Med 2009, 50:1801-1807.
237. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, Okubo Y,
Suhara T: Perifere benzodiazepinreceptorer hos patienter med kroniske
skizofreni: et PET-studie med [11C]DAA1106. Int J
Neuropsychopharmacol 2010, 13:943-950.
238. Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B,
Spellmann I, Hetzel G, Maino K, Kleindienst N, Müller HJ, Arolt V, Riedel M:
Cyclooxygenase-2-hæmmeren celecoxib har terapeutiske virkninger i
svær depression: resultater af en dobbeltblind, randomiseret placebo
kontrolleret, add-on pilotundersøgelse til reboxetin. Mol Psykiatri 2006,
11: 680 684.
239. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, MohebbiRasa
S, Raznahan M, Kamalipour A: Klinisk forsøg med supplerende celecoxib
behandling hos patienter med svær depression: en dobbeltblind og
placebokontrolleret forsøg. Depress Anxiety 2009, 26:607�611.
240. Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N:
Forkortet virkningsstart af antidepressiva ved svær depression vha
acetylsalicylsyreforøgelse: et åbent pilotstudie. Int Clin
Psychopharmacol 2006, 21:227-231.
241. Uher R, Carver S, Power RA, Mors O, Maier W, Rietschel M, Hauser J,
Dernovsek MZ, Henigsberg N, Souery D, Placentino A, Farmer A, McGuffin P:
Ikke-steroide antiinflammatoriske lægemidler og effektiviteten af antidepressiva i
svær depressiv lidelse. Psychol Med 2012, 42:2027-2035.
242. Müller N, Riedel M, Scheppach C, Brandstatter B, Sokullu S, Krampe K,
Ulmschneider M, Engel RR, Møller HJ, Schwarz MJ: gavnligt antipsykotisk middel.
effekter af celecoxib-tillægsbehandling sammenlignet med risperidon alene i
skizofreni. Am J Psychiatry 2002, 159:1029-1034.
243. Müller N, Riedel M, Schwarz MJ, Engel RR: Kliniske virkninger af COX-2
inhibitorer på kognition ved skizofreni. Eur Arch Psychiatry Clin Neurosci
2005, 255:149�151.
244. Müller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M,
Møller HJ, Klauss V, Schwarz MJ, Riedel M: Celecoxib-behandling i en tidlig
stadium af skizofreni: resultater af en randomiseret, dobbeltblind, placebokontrolleret
forsøg med celecoxib-forøgelse af amisulpridbehandling.
Schizophr Res 2010, 121:118-124.
245. Sayyah M, Boostani H, Pakseresht S, Malayeri A: En foreløbig randomiseret
dobbeltblindet klinisk forsøg på effektiviteten af celecoxib som et supplement til
behandling af obsessiv-kompulsiv lidelse. Psychiatry Res 2011,
189: 403 406.
246. Sublette ME, Ellis SP, Geant AL, Mann JJ: Meta-analyse af virkningerne af
eicosapentaensyre (EPA) i kliniske forsøg med depression. J Clin
Psychiatry 2011, 72:1577�1584.
247. Bloch MH, Hannestad J: Omega-3 fedtsyrer til behandling af
depression: systematisk gennemgang og meta-analyse. Mol Psykiatri 2012,
17: 1272 1282.
248. Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL: A
gennemgang af anti-inflammatoriske midler til symptomer på skizofreni.
J Psychopharmacol.
249. Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P:
Antidepressive virkninger af selektive serotoningenoptagshæmmere (SSRI'er)
svækkes af antiinflammatoriske lægemidler hos mus og mennesker. Proc Natl
Acad Sci USA 2011, 108:9262�9267.
250. Gallagher PJ, Castro V, Fava M, Weilburg JB, Murphy SN, Gainer VS, Churchill
SE, Kohane IS, Iosifescu DV, Smoller JW, Perlis RH: Antidepressiv respons
hos patienter med svær depression udsat for NSAID'er: a
lægemiddelovervågning. Am J Psychiatry 2012, 169:1065-1072.
251. Shelton RC: Nedsætter samtidig brug af NSAID'er effektiviteten af
antidepressiva? Am J Psychiatry 2012, 169:1012-1015.
252. Martinez-Gras I, Perez-Nievas BG, Garcia-Bueno B, Madrigal JL, AndresEsteban
E, Rodriguez-Jimenez R, Hoenicka J, Palomo T, Rubio G, Leza JC:
Det antiinflammatoriske prostaglandin 15d-PGJ2 og dets nukleare receptor
PPARgamma er nedsat ved skizofreni. Schizophr Res 2011,
128: 15 22.
253. Garcia-Bueno B, Perez-Nievas BG, Leza JC: Er der en rolle for atomkraftværket
receptor PPARgamma i neuropsykiatriske sygdomme? Int J
Neuropsychopharmacol 2010, 13:1411-1429.
254. Meyer U: Anti-inflammatorisk signalering ved skizofreni. Hjernens adfærd
Immun 2011, 25:1507-1518.
255. Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M,
Hinz B: COX-2 og PPAR-gamma giver cannabidiol-induceret apoptose
af humane lungekræftceller. Mol Cancer Ther 2013, 12:69�82.
256. Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF,
Godbout JP: Minocyclin dæmper lipopolysaccharid (LPS)-induceret
neuroinflammation, sygdomsadfærd og anhedoni.
J Neuroinflammation 2008, 5:15.
257. Sarris J, Mischoulon D, Schweitzer I: Omega-3 til bipolar lidelse: metaanalyser
til brug ved mani og bipolar depression. J Clin Psychiatry 2012,
73: 81 86.
258. Amminger GP, Schafer MR, Papageorgiou K, Klier CM, Cotton SM, Harrigan
SM, Mackinnon A, McGorry PD, Berger GE: Langkædede omega-3 fedtsyrer
til indiceret forebyggelse af psykotiske lidelser: en randomiseret, placebokontrolleret
forsøg. Arch Gen Psychiatry 2010, 67:146-154.
259. Fusar-Poli P, Berger G: Eicosapentaensyreinterventioner i
skizofreni: meta-analyse af randomiserede, placebo-kontrollerede undersøgelser.
J Clin Psychopharmacol 2012, 32:179-185.
260. Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S: Neurosteroider,
stress og depression: Potentielle terapeutiske muligheder.
Neurosci Biobehav Rev 2013, 37:109-122.
261. Uhde TW, Singareddy R: Biologisk forskning i angstlidelser. I
Psykiatri som neurovidenskab. Redigeret af Juan Jose LI, Wolfgang G, Mario M,
Norman S. Chichester: John Wiley & Sons, Ltd; 2002:237�286.
262. Gibson SA, Korado Z, Shelton RC: Oxidativ stress og glutathion
respons i vævskulturer fra personer med svær depression.
J Psychiatr Res 2012, 46:1326-1332.
263. Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN,
Bowden CL, Soares JC: Celecoxib som et supplement i behandlingen af
depressive eller blandede episoder af bipolar lidelse: en dobbeltblind,
randomiseret, placebokontrolleret undersøgelse. 2008, 23:87�94.
264. Levine J, Cholestoy A, Zimmerman J: Mulig antidepressiv effekt af
minocyclin. 1996, 153:582.
265. Levkovitz Y, Mendlovich S, Riwkes S, Braw Y, Levkovitch-Verbin H, Gal G,
Fennig S, Treves I, Kron S: En dobbeltblind, randomiseret undersøgelse af
minocyclin til behandling af negative og kognitive symptomer i
tidlig fase skizopreni. J Clin Psychiatry 2010, 71:138-149.
266. Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Mulig antipsykotisk effekt af minocyclin hos patienter med
skizofreni. Prog Neuropsychopharmacol Biol Psychiatry 2007, 31:304-307.
267. Miyaoka J, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J:
Minocyclin som supplerende terapi for skizofreni: en åben etiket
undersøgelse. 2008, 31:287�292.
268. Rodriguez CI, Bender Jr., Marcus SM, Snape M, Rynn M, Simpson HB:
Minocyklinforøgelse af farmakoterapi i tvangs-kompulsiv
lidelse: et åbent forsøg. 2010, 71:1247�1249.
doi:10.1186/1742-2094-10-43
Citer denne artikel som: Najjar et al.: Neuroinflammation and psychiatric
sygdom. Journal of Neuroinflammation 2013 10:43.
Luk harmonika
Oplysningerne heri om "Neuroinflammation og psykiatrisk sygdom" er ikke beregnet til at erstatte et en-til-en-forhold med en kvalificeret sundhedsperson eller autoriseret læge og er ikke medicinsk rådgivning. Vi opfordrer dig til at træffe sundhedsbeslutninger baseret på din forskning og partnerskab med en kvalificeret sundhedsperson.
Bloginformation og diskussioner om omfang
Vores informationsomfang er begrænset til kiropraktik, muskuloskeletal, fysisk medicin, wellness, bidragende ætiologisk viscerosomatiske forstyrrelser inden for kliniske præsentationer, tilhørende somatovisceral refleks klinisk dynamik, subluksationskomplekser, følsomme helbredsproblemer og/eller funktionel medicin artikler, emner og diskussioner.
Vi giver og præsenterer klinisk samarbejde med specialister fra forskellige discipliner. Hver specialist er styret af deres faglige omfang af praksis og deres licensjurisdiktion. Vi bruger funktionelle sundheds- og velværeprotokoller til at behandle og understøtte pleje af skader eller lidelser i bevægeapparatet.
Vores videoer, indlæg, emner, emner og indsigt dækker kliniske forhold, problemstillinger og emner, der relaterer til og direkte eller indirekte understøtter vores kliniske anvendelsesområde.*
Vores kontor har med rimelighed forsøgt at give støttende citater og har identificeret den eller de relevante forskningsundersøgelser, der understøtter vores indlæg. Vi leverer kopier af understøttende forskningsundersøgelser tilgængelige for tilsynsråd og offentligheden efter anmodning.
Vi forstår, at vi dækker forhold, der kræver yderligere forklaring på, hvordan det kan hjælpe med en bestemt plejeplan eller behandlingsprotokol. derfor er du velkommen til at spørge for yderligere at diskutere emnet ovenfor Dr. Alex Jimenez, DC, eller kontakte os på 915-850-0900.
Vi er her for at hjælpe dig og din familie.
Blessings
Dr. Alex Jimenez A.D. MSACP, RN*, CCST, Ifmcp*, CIFM*, ATN*
Email: coach@elpasofunctionalmedicine.com
Licenseret som Doctor of Chiropractic (DC) i Texas & New Mexico*
Texas DC-licensnummer TX5807, New Mexico DC Licensnr. NM-DC2182
Licenseret som registreret sygeplejerske (RN*) in Florida
Florida-licens RN-licens # RN9617241 (Kontrol nr. 3558029)
Kompakt status: Multi-State Licens: Bemyndiget til at praktisere i 40 stater*
Dr. Alex Jimenez DC, MSACP, RN*CIFM*, IFMCP*, ATN*, CCST
Mit digitale visitkort





